7.3. Control the Cluster Shape
Tip
The sample input and output files can be found in testfiles/geom/1-shape.
In this Section we will introduce how to use components block to accurately control the cluster shape. You will see how powerful geom is.
7.3.1. General Input
In this section we will use the same input for all examples, only changing the lm_dir and components block.
1lm_dir TODO # Save the local minima to this folder.
2num_calcs 10 # Total number of calculations.
3do_coarse_opt yes # no: Do NOT the coarse optimization.
4min_energy_gap 1.E-4 # When two energies differ smaller than
5 # this value, they are treated as identical.
6 # A negative number means do not remove
7 # energetically degenerated ones.
8max_geom_iters 3000 # The maximum number of iterations for local optimization.
9 # If it is less or equal than zero, then the number is unlimited.
10
11components
12 TODO
13end
14
15commands
16end
You can see that, compared with the one given in Input File for geom, the charmm, xtb, and savegjf are deleted, since we do not need them. Also, the commands block is left empty, since we do not need any third party programs. We just use geom as a structure generator. For each system, we generate only 10 clusters, as indicated by num_calcs.
7.3.2. Example: box, (C6H6)512(H2O)2048
We will consider the cluster of 512 \(\mathrm{C}_6\mathrm{H}_6\) and 2048 \(\mathrm{H}_2\mathrm{O}\). First we prepare their structure:
112
2C6H6
3C 0.00000000 1.39649000 0.00000000
4C 1.20939600 0.69824500 0.00000000
5C 1.20939600 -0.69824500 0.00000000
6C 0.00000000 -1.39649000 0.00000000
7C -1.20939600 -0.69824500 0.00000000
8C -1.20939600 0.69824500 0.00000000
9H 0.00000000 2.48319100 0.00000000
10H 2.15050600 1.24159500 0.00000000
11H 2.15050600 -1.24159500 0.00000000
12H 0.00000000 -2.48319100 0.00000000
13H -2.15050600 -1.24159500 0.00000000
14H -2.15050600 1.24159500 0.00000000
13
2water
3O 0.00000000 0.00000000 -0.11081188
4H 0.00000000 -0.78397589 0.44324751
5H -0.00000000 0.78397589 0.44324751
7.3.2.1. Mixture
First, we want to get a solvent box of their mixture. Here, the keyword box will be used. In h2o-c6h6-mixture.inp, change the component block:
1lm_dir h2o-c6h6-mixture
2components
3 c6h6.xyz 512
4 box 0 0 0 45 90 45
5 ****
6 h2o.xyz 2048
7 box 0 0 0 45 90 45
8 ****
9end
Here we can see that:
Each component is separated by
****;The first line of each component is the structure file name (path can be included) and its number. Say
h2o.xyz 2048means that there are2048water, whose structure is defined inh2o.xyz.The second line of each component is its shape of packing.
In this case, both components have the shape box:
Note
box x0 y0 z0 x1 y1 z1
Uniformly distribute molecules in a box determined by (x0, y0, z0) and (x1, y1, z1).
Since \(\mathrm{C}_6\mathrm{H}_6\) and \(\mathrm{H}_2\mathrm{O}\) are distributed in the same space, they will form a perfect mixture.
Now run the program:
$ geom h2o-c6h6-mixture.inp > h2o-c6h6-mixture.out
7.3.2.2. Bi-phase
Since \(\mathrm{C}_6\mathrm{H}_6\) and \(\mathrm{H}_2\mathrm{O}\) do not solvate each other, it is possible to generate a bi-phase cluster. Just let them distribute in different but adjoint boxes!
1lm_dir h2o-c6h6-biphase
2components
3 c6h6.xyz 512
4 box 0 0 0 45 45 45
5 ****
6 h2o.xyz 2048
7 box 0 45 0 45 90 45
8 ****
9end
Now run the program:
$ geom h2o-c6h6-biphase.inp > h2o-c6h6-biphase.out
Now, let us compare the structures. Just randomly take 2, say h2o-c6h6-mixture/3.xyz and h2o-c6h6-biphase/2.xyz. See below, we perfectly get what we want!

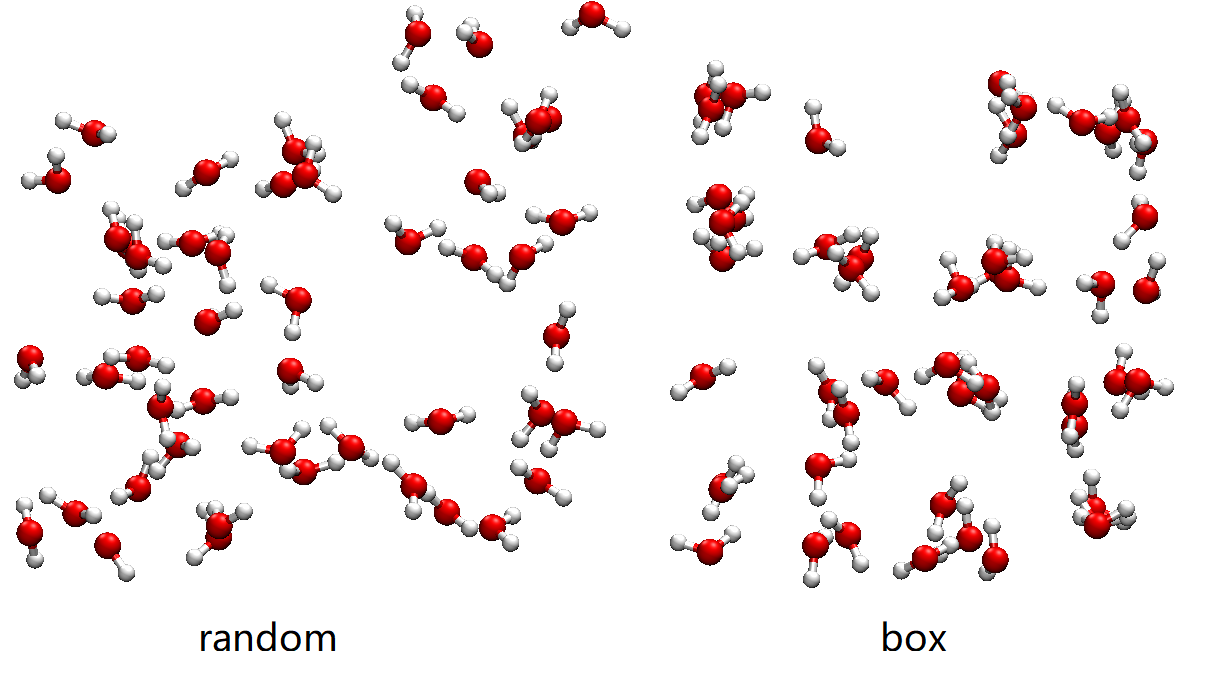
7.3.3. Example: random, (H2O)50
There is a keyword random that is very similar to box:
Note
random x0 y0 z0 x1 y1 z1
Randomly distribute molecules in a region determined by (x0, y0, z0) and (x1, y1, z1).
Unlike box, which tend to put molecules uniformly, random will put molecules randomly. Let’s try it for 50 water molecules.
1lm_dir h2o50
2components
3 h2o.xyz 50
4 random 0 0 0 15 15 15
5 ****
6end
Now run the program:
$ geom h2o50.inp > h2o50.out
One structure, say h2o50/3.xyz, is shown below. We compared it with a structure generated by replacing random to box.
7.3.4. Example: fix, H2O in SSZ-13
We want to study the diffusion of \(\text{H}_2\text{O}\) in zeolite SSZ-13. The structure of SSZ-13 is given below:
1108
2SSZ-13
3 O 5.7000232468 7.6303196339 2.6186286767
4 O 5.2045498989 10.1995252965 3.2272552812
5 O 7.6341828896 9.4099723840 2.7745789292
6 O 5.8753034436 9.5497196844 0.7672162010
7 O 7.6375473635 4.2759815544 2.6753670218
8 O 9.5707533006 7.6299616532 2.6176480074
9 O 9.6106668261 5.3414448452 13.6228779840
10 O 7.6185326402 8.7535796994 13.6836873966
11 O 5.6658146587 5.3461814248 13.6160993667
12 O 5.7026893239 -0.2642085908 7.5407946669
13 O 0.7825962064 8.1512328930 7.5378038833
14 O 2.7836661828 11.6286830319 7.5359961843
15 O 2.7363955771 9.3420684719 3.8535391861
16 O 7.6423184053 0.8505970745 3.7730132707
17 O 12.5335364638 9.3415768142 3.8544585628
18 O 12.4624155643 3.6818551888 12.4626958554
19 O 7.6062858624 12.1072824025 12.4587051589
20 O 2.8065149113 3.6894351696 12.2320710199
21 O 2.7322963802 1.4435907562 8.8508132419
22 O 0.8127191930 4.8681935127 8.6967338458
23 O 12.4822455137 1.3976417344 8.7679437480
24 O 5.6527918209 2.5629377561 3.2319866240
25 O 12.0423876809 6.7757052402 3.2233313473
26 O 12.0554980341 6.2591391560 13.0723873245
27 O 5.5916803863 10.4417381114 13.0790389503
28 O 5.1667070765 2.7598543540 13.1500955201
29 O 10.0234139879 2.7573797734 13.0802094325
30 O 9.6173269693 10.4509455156 13.0839569435
31 O 3.1861034312 6.1951039344 13.1391153838
32 O 3.2262073712 6.7771124228 3.2237580685
33 O 9.6209190582 2.5656089905 3.2277416465
34 O 10.0640705757 10.1974856514 3.2288444611
35 O 5.1427255382 2.3096527675 8.0673522413
36 O 12.4246061731 6.5049032168 8.1596286214
37 O 5.2287254916 10.6584161272 8.1499333643
38 O 10.0660760251 10.7260767207 8.1486535093
39 O 2.8490782073 6.5139838953 8.1476193260
40 O 10.0374817601 2.3659377558 8.1472483774
41 O 5.1316112190 5.0641064531 2.8432015310
42 O 10.1446584873 5.0657040841 2.7757616149
43 O 10.1029249905 7.9248177185 13.5271012901
44 O 5.1139616931 7.9307810948 13.5327555244
45 O 7.6221919820 3.6097600857 13.5295025529
46 O 7.6288414056 1.5118665409 7.7615136197
47 O 11.9654721427 9.0151964046 7.6848839357
48 O 3.2451981211 9.0440458535 7.6881718795
49 O 3.3069973094 11.9100887748 3.6976804332
50 O 5.1307773743 0.0649280529 3.6340011283
51 O 0.7969499830 7.5670851250 3.6935855226
52 O 0.7910818700 5.3346368625 12.6820331150
53 O 11.9636291062 1.1105277839 12.6111708865
54 O 3.2392803629 1.1213561083 12.5479345449
55 O 3.3075057508 4.0157430778 8.7760041016
56 O 12.0156624591 3.9738388657 8.6139906116
57 O 7.6320672727 11.5168215978 8.6125370060
58 O 5.8866593170 3.4930627815 0.7891245303
59 O 11.1355336190 6.5074683788 0.7610373808
60 O 9.3801650379 3.4821683317 0.7717307825
61 O 9.3883869933 9.5474036098 0.7666774122
62 O 4.1692472661 6.4488991333 0.7780060769
63 O 5.9556046254 1.6428018426 5.6765493427
64 O 12.6861939240 7.3729754052 5.6887347990
65 O 4.3586450055 10.4595476971 5.6921261702
66 O 2.5801899718 7.3759353889 5.6905185401
67 O 9.3400531831 1.7041165806 5.6889457138
68 O 10.9432024258 10.5171940688 5.6851549584
69 O 12.7365486472 5.5973335861 10.6155686479
70 O 5.8478045129 11.3130579735 10.6124834850
71 O 4.5150336200 2.4391618565 10.6065434296
72 O 9.3801870229 11.2550644389 10.6124530044
73 O 2.4362265224 5.7858392934 10.6232169362
74 O 10.9023169714 2.5612785425 10.6093785249
75 Si 6.0920814665 3.8508163034 2.3648358150
76 Si 10.7202283849 6.5092425635 2.3328070616
77 Si 10.7198656965 6.4925660943 13.9546031759
78 Si 6.0652860896 9.1709124986 13.9647009255
79 Si 6.0809445096 3.8112454238 13.9744263842
80 Si 9.1680499571 3.8090892108 13.9600924915
81 Si 9.1702883031 9.1714808622 13.9670464697
82 Si 4.5450589507 6.4778439590 13.9665556825
83 Si 4.5589301028 6.4970522597 2.3544741665
84 Si 9.1776796571 3.8440220779 2.3459143784
85 Si 9.1743108787 9.1858945424 2.3361918038
86 Si 6.1023939667 1.2912554713 7.2563853198
87 Si 12.8984444715 7.7609462493 7.2559239768
88 Si 3.8937893347 10.4628352540 7.2542014120
89 Si 3.8917972712 10.4639571217 4.1313484718
90 Si 6.1021660505 1.2768708902 4.0983248150
91 Si 12.9264443751 7.7793779850 4.1294213351
92 Si 2.3418034349 7.7806749736 4.1302867232
93 Si 9.1747023738 1.2939288098 4.1186514298
94 Si 11.3852128233 10.4740999990 4.1168223128
95 Si 11.3849847412 10.4645367999 7.2520286806
96 Si 2.3463927644 7.7693527709 7.2545905923
97 Si 9.1641901801 1.3288002236 7.2655864551
98 Si 12.9252078158 5.2179055842 12.1892578243
99 Si 6.0541114995 11.7025286295 12.1775311953
100 Si 12.9318493098 5.2383696707 9.0377621396
101 Si 6.0833435431 11.6953330771 9.0489768990
102 Si 9.1739017270 11.7144997047 9.0639602124
103 Si 2.3459232172 5.2847394268 9.0735577386
104 Si 11.3678806369 2.5575039972 9.0491343651
105 Si 11.3433313927 2.5385553857 12.1747149889
106 Si 9.1561829214 11.6952711342 12.1629072761
107 Si 2.3228901646 5.2436972032 12.1586452445
108 Si 6.0937069986 9.1867058586 2.3360857945
109 Si 3.9151902179 2.5466565165 9.0895664436
110 Si 3.9346333440 2.5195603195 12.1307588181
In this case, SSZ-13 should be fixed in space, and water molecules are put into its big cage. In this case, a new keyword fix will help:
Note
fix x y z
Put the center of the molecule at (x, y, z), with a random rotation.
fix x y z alpha beta gamma
Put the center of the molecule at (x, y, z), with a rotation (alpha, beta, gamma). So, fix x y z 0 0 0 will not rotate the molecule but keep it orientation as in its XYZ file.
We fix the center of SSZ-13 at (0, 0, 0) without any rotations, so:
1lm_dir h2o-ssz-13
2components
3 ssz-13.xyz 1
4 fix 0 0 0 0 0 0
5 ****
6end
After running the optimization, you will find that SSZ-13 have been moved that its center is at the origin without rotations. Now we can try to determine a box inside the cage to let us put water in.
Tip
Their are a lot of ways to do this. One way is to use VMD. Load the optimized structure, say h2o-ssz-13/0.xyz to VMD, and in its command line, run the following command:
vmd > measure minmax [atomselect top all]
{-6.468430519104004 -6.547443389892578 -6.980508804321289} {5.680822849273682 5.824047565460205 6.232880115509033}
This {-6.468430519104004 -6.547443389892578 -6.980508804321289} {5.680822849273682 5.824047565460205 6.232880115509033} is the box that contains the whole system. Of course, it should be smaller, so we shrink it to, say {-2 -3 -4} {2 3 4}.
Now, we can prepare the complete input file:
1lm_dir h2o-ssz-13
2components
3 ssz-13.xyz 1
4 fix 0 0 0 0 0 0
5 ****
6 h2o.xyz 10
7 random -2 -3 -4 2 3 4
8 ****
9end
Run the program:
$ geom h2o-ssz-13.inp > h2o-ssz-13.out
Let’s check out a structure, say h2o-ssz-13/3.xyz, which is shown below. You can use this as an initial guess for first-principles calculation!

7.3.5. Example: droplet, NaCl in water droplet
We want to study 3 \(\text{Na}^{+}\) and \(\text{Cl}^{-}\) in a water microdroplet. The structures of \(\text{Na}^{+}\) and \(\text{Cl}^{-}\) are shown below.
11
2Na
3Na 0 0 0
11
2Cl
3Cl 0 0 0
To distribute the species in a droplet, we use the keyword droplet.
Note
droplet x y z a b c
Randomly distribute molecules in an ellipsoid centered at (x, y, z) with 3 radii a, b, and c.
So we can distribute them in the same ellipsoid (say, three radii being 20, 10, and 10) to model the solvation:
1lm_dir h2o-na3cl3
2components
3 h2o.xyz 600
4 droplet 0 0 0 20 10 10
5 ****
6 na.xyz 3
7 droplet 0 0 0 20 10 10
8 ****
9 cl.xyz 3
10 droplet 0 0 0 20 10 10
11 ****
12end
Run the program:
$ geom h2o-na3cl3.inp > h2o-na3cl3.out
Let’s check out a structure, say h2o-na3cl3/6.xyz, which is shown below.

7.3.6. Example: absorb, (CO)10@Ag38
There is a very useful keyword absorb in geom: with it you can put molecules on the surface of a molecule, like a metal slab or protein.
Note
absorb n1 n2 n3
Randomly distribute molecules on the surface of component n1 (index starting from 1). These molecules take a direction that the vector from atom n2 to n3 points along the local normal.
Let’s show this with an example. Consider 10 \(\text{CO}\) absorbed on \(\text{Ag}_{38}\). The structures of \(\text{Ag}_{38}\) and \(\text{CO}\) are given below:
138
2Ag38
3Ag 1.83581324854909 -0.66521416103382 -4.05084800386537
4Ag 3.26146602865993 1.43076353307629 -2.74559379561526
5Ag -1.86766331981303 -3.69246865502893 1.76047772000301
6Ag -1.83581102159633 0.66521241553523 4.05084656236035
7Ag -4.37635842854696 0.46053090761453 0.92628644673458
8Ag 0.15602085746999 2.00172223612582 -0.25803332624779
9Ag 4.09045041549422 -0.07826673249061 1.86662431725502
10Ag 2.96684423722295 -2.41279942319018 0.63392161141815
11Ag 1.27537017794612 0.10273478583599 1.56858140117429
12Ag 0.49180992899154 1.78611199051730 -3.40510826327950
13Ag -1.27542752460590 -0.10273282991459 -1.56852780260341
14Ag 1.86765806332299 3.69248093139238 -1.76047676908242
15Ag 1.58003106702717 4.07712387902652 1.04989147026445
16Ag 1.24797585452742 -4.25706785477554 -0.73581396857223
17Ag -0.97472878274367 -0.48635742691014 -4.36290889209247
18Ag 1.56411023996142 -0.28340514372109 -1.25313947728674
19Ag -3.31177359770293 -2.01320436495574 -0.06340730066674
20Ag 0.96033695393650 -3.87241791982209 2.07453413622748
21Ag -0.49179643241507 -1.78609168703799 3.40506272467468
22Ag 0.97471867683167 0.48635633328589 4.36290696130676
23Ag 4.37636506050101 -0.46053366775522 -0.92629395693753
24Ag 2.95063571629445 -2.55651271910380 -2.23163152893035
25Ag -1.24798342397203 4.25705941555808 0.73581747916760
26Ag -2.37713910520774 1.78950388800175 -3.37174214960943
27Ag 2.37713234562125 -1.78950289396663 3.37174836642454
28Ag 3.31183702084341 2.01323876759066 0.06340286526436
29Ag -0.96034478186356 3.87241695907278 -2.07452887108150
30Ag -3.26146124524853 -1.43076585076549 2.74558710274998
31Ag -2.95063365988223 2.55651250079272 2.23162668919665
32Ag -1.58003774094824 -4.07711813992677 -1.04988736473955
33Ag -2.68795036663320 -2.19763079149194 -2.85783568469868
34Ag -4.09044728739259 0.07826707984227 -1.86660956451639
35Ag -0.14676607800026 2.64016188401185 2.83464031184915
36Ag 0.14677416984600 -2.64018472712655 -2.83466944323257
37Ag -0.15608921802641 -2.00170980842979 0.25806971579687
38Ag 2.68794654621046 2.19763200878331 2.85784466194340
39Ag -2.96679571349412 2.41276479806487 -0.63391785806270
40Ag -1.56408890116479 0.28339049331867 1.25310346730934
12
2CO
3C 0.00000000000000 0.00000000000000 -0.06362328061977
4O 0.00000000000000 0.00000000000000 1.06362328061977
Now we prepare the input:
1lm_dir ag38-co10
2components
3 ag38.xyz 1
4 fix 0 0 0 0 0 0
5 ****
6 co.xyz 10
7 absorb 1 1 2
8 ****
9end
First, we simply fix \(\text{Ag}_{38}\) as (0, 0, 0) without rotations, then let 10 \(\text{CO}\) absorb on the 1``st component, with the vector formed by the ``1``st and ``2``nd atom of ``co.xyz pointing outward. Let’s run the optimization:
Run the program:
$ geom ag38-co10.inp > ag38-co10.out
Let’s check out a structure, say ag38-co10/3.xyz, which is shown below.

If you change absorb 1 1 2 to absorb 1 2 1, you will see that oxygen atoms of \(\text{CO}\) will contact with the metal slab surface.
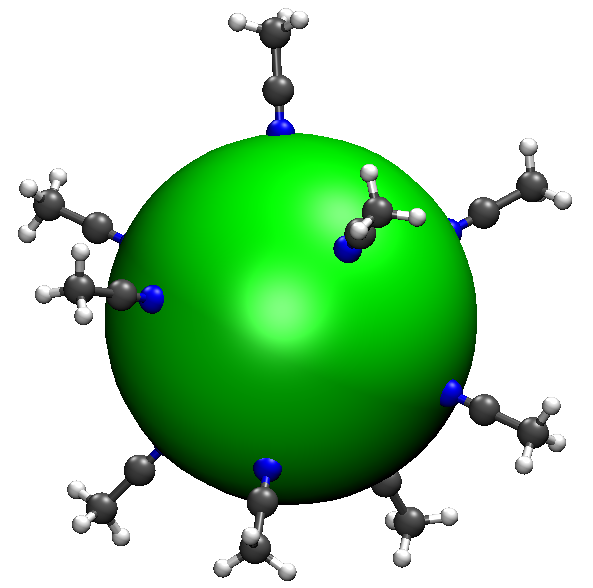
7.3.7. Example: shell, (CH3CN)10
Note
shell n1 n2 x y z a b c
Uniformly distribute molecules over an ellipsoid centered at (x, y, z) with 3 radii a, b, and c. These molecules take a direction that the vector from atom n1 to n2 points along the local normal.
shell n1 n2 n3 n4 x y z a b c
Uniformly distribute molecules over an ellipsoid centered at (x, y, z) with 3 radii a, b, and c. These molecules take a direction that the vector from the center of n1, n2 to the center of n3, n4 points along the local normal.
shell n1 n2 n3 n4 n5 n6 x y z a b c
Uniformly distribute molecules over an ellipsoid centered at (x, y, z) with 3 radii a, b, and c. These molecules take a direction that the vector from the center of n1, n2, n3 to the center of n4, n5, n6 points along the local normal.
shell is a powerful keyword in geom which enables you to build:
Micelle, anti-micelle structures;
Ligated clusters.
These two applications will be discussed in this and following sections. In this section, we will show how to let 10 \(\text{CH}_3\text{CN}\) uniformly distribute over a sphere.
The structure of \(\text{CH}_3\text{CN}\) is given below:
16
2CH3CN
3C 0.00000000 0.00000000 -1.22619100
4H 0.00000000 1.00880600 -1.58285700
5H -0.87365100 -0.50440300 -1.58285700
6H 0.87365100 -0.50440300 -1.58285700
7C 0.00000000 0.00000000 0.31380900
8N 0.00000000 0.00000000 1.46040900
Use the following input:
1lm_dir ch3cn10
2components
3 ch3cn.xyz 10
4 shell 6 5 0 0 0 5 5 5
5 # Or you can use the following:
6 # shell 6 6 6 2 3 4 0 0 0 5 5 5
7 ****
8end
Here, 0 0 0 5 5 5 means that molecules will be distributed over the ellipsoid centered at (0, 0, 0) with 3 radii 5, 5, 5 (so it is a sphere). In the first way, 6 5 means that atom 6 is closer to the ellipsoid, and 6 5 points along the local normal; in the second way, 6 6 6 2 3 4 means that the center of atom 6 6 6 (i.e. the nitrogen atom) is closer to the ellipsoid, and the center of 2 3 4 (i.e. the three hydrogen atoms) is pointing outwardly. The two ways have the same effect.
Run the program:
$ geom ch3cn10.inp > ch3cn10.out
Let’s check out a structure, say ch3cn10/6.xyz, which is shown below.
7.3.8. Example: layer, (C4H2)30
layer can let molecules for a layer. This is very useful for building films, membranes, etc.
Note
layer n1 n2 n3 x0 y0 z0 x1 y1 z1 x2 y2 z2 x3 y 3 z 4
A finite layer is determined by 3 points: (x1, y1, z1), (x2, y2, z2), and (x3, y3, z3). Molecules will be distributed on the plane with a direction that the vector from atom n2 to n3 points along the local normal (x0, y0, z0). If n1 is 1, molecules will be distributed regularly; if it is 2, then distributed randomly.
For example, we want to arrange 30 \(\text{C}_4\text{H}_2\) to form a 5x5x5 film. The structure of \(\text{C}_4\text{H}_2\) is:
16
2C4H2
3C -0.00000000 -0.00000000 -1.97120000
4H -0.00000000 -0.00000000 -3.04120000
5C -0.00000000 -0.00000000 -0.77000000
6C 0.00000000 -0.00000000 0.77000000
7C 0.00000000 -0.00000000 1.97120000
8H 0.00000000 -0.00000000 3.04120000
This is a linear molecule. The following input can be used:
1lm_dir c4h230
2components
3 c4h2.xyz 30
4 layer 1 2 1 0 0 1 5 0 0 5 5 0 0 5 0
5 ****
6end
Here, 5 0 0 5 5 0 0 5 0 defines a layer of (5, 0, 0), (5, 5, 0), and (0, 5, 0). 2 1 means that atom 2 is closer to the layer, and 2 1 points along the local normal (0, 0, 1),. The first 1 means they are distributed regularly.
Run the program:
$ geom c4h230.inp > c4h230.out
Let’s check out a structure, say c4h230/3.xyz, which is shown below.

You can try to see what you will get by changing 2 1 1 to 2 1 2.