7.8. geom with VASP
Tip
VASP is useful for clusters in periodic systems, like surface-supported, or zeolite-absorbed systems. You’d better have some experience of condensed-phase physics or solid quantum chemistry before reading this section.
To use VASP with geom, you need two files: misc/runVASP.py.
misc/runVASP.py is a script to call VASP. There are ONLY 2 things that you need to change:
1vaspcmd = "vasp_std"
2pbc = [[8.1426336762816440, 0.0000000000000000, 0.0000000000000000],
3 [-4.0677955560985355, 7.0495917923679352, 0.0000000000000000],
4 [0.0000000000000000, 0.0000000000000000, 30.0000000000000000]]
You need to change vaspcmd to the correct VASP calling command on your computer, and pbc to the cell sizes of your system.
As usual, you should prepare INCAR, POTCAR, and KPOINTS for running VASP calculations.
7.8.1. Example: Se3 on C2N
Tip
The sample input and output files can be found in testfiles/geom/11-c2n-vasp.
Let’s see how 3 \(\text{Se}\) are distributed over the surface of \(\text{C}_2\text{N}\). The structure of \(\text{C}_2\text{N}\) is
118
2C2N
3N 0.64116352 3.56117427 5.67809614
4N 2.69450515 2.34860406 5.67308619
5N 2.71754532 4.73169504 5.67345986
6N 5.41920549 2.39517656 5.67702623
7N 5.39621102 0.01223017 5.67804973
8N -0.67007960 1.18256295 5.67743531
9C 1.31336867 2.33226424 5.68025714
10C 1.32380529 4.76901081 5.68336725
11C 3.42905165 3.53718653 5.67716633
12C 4.68474934 1.20655351 5.67969992
13C 6.78995796 -0.02520025 5.68329936
14C -1.34242672 2.41165268 5.68568288
15C 4.79530371 3.61561247 5.68882985
16C 3.31831615 1.12807079 5.68711514
17C 0.69185103 1.10770299 5.68930373
18C -0.72092338 3.63609485 5.69624406
19C 3.47041412 5.87283681 5.69021702
20C 0.57555247 5.92048738 5.69537016
This is the Cartesian coordinates of \(\text{C}_2\text{N}\). If you add cell sizes to build a POSCAR and visualize it, the structure is like below:
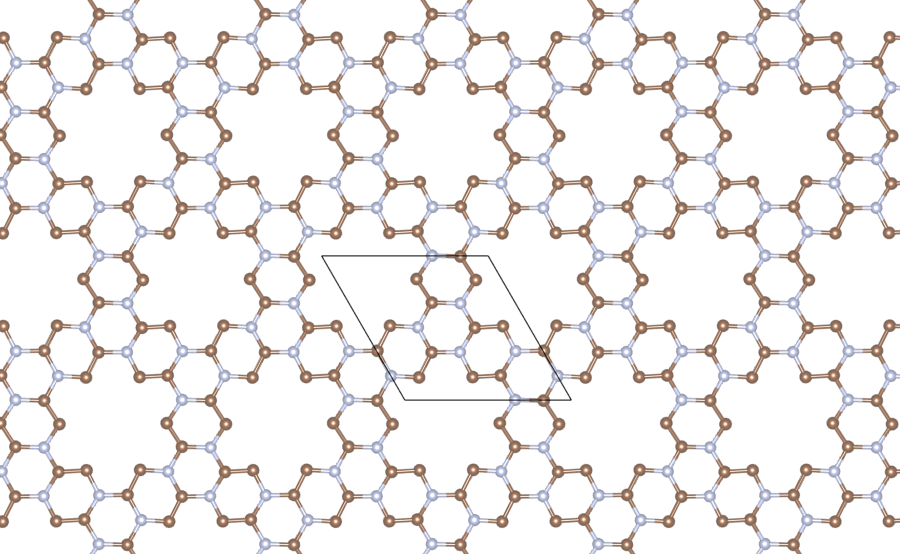
The selenium atom is:
11
2Se
3Se 0 0 0
The input file is:
1lm_dir c2nse3 # Save the local minima to this folder.
2num_calcs 20 # Total number of calculations.
3do_coarse_opt yes # no: Do NOT the coarse optimization.
4min_energy_gap 1.E-4 # When two energies differ smaller than
5 # this value, they are treated as identical.
6 # A negative number means do not remove
7 # energetically degenerated ones.
8max_geom_iters 3000 # The maximum number of iterations for local optimization.
9 # If it is less or equal than zero, then the number is unlimited.
10
11components
12 c2n.xyz 1
13 fix 0 0 5 0 0 0
14 ****
15 se.xyz 3
16 random 0 0 6 6 6 8
17 ****
18end
19
20commands
21python runVASP.py $inp$ $out$ $xxx$
22end
In this input file, we fix \(\text{C}_2\text{N}\) at (0, 0, 5) without rotations, and let the selenium atoms distribute randomly above the surface.
Make sure in the current directory you have prepared runVASP.py, INCAR, POTCAR, and KPOINTS.
Now you can run the global optimization:
$ geom c2nse3.inp > c2nse3.out
This is a quite long job. For me, it took 9 days. After the optimization, the end of c2nse3.out is
-- Result Report --
Results are energy-increasingly reordered.
Structures of energies within 1.000E-04 are treated as degenerate.
All minima are saved to "c2nse3".
-------------------------------------------------------------------
# index Energy NaiveRMSD
-------------------------------------------------------------------
0 17 -159.07404300 0.00000000
1 2 -158.74814600 1.24247428
2 7 -158.74802600 2.12024945
3 16 -158.74768300 3.16425048
4 14 -158.73993100 3.01598380
5 15 -158.73965600 2.06890365
6 19 -158.72750400 2.06285077
7 13 -158.72533900 1.12698497
8 9 -157.98449500 3.90976227
The first one, c2nse3/17.xyz, seems to have a too low energy compared with others, and c2nse3/2.xyz seems to be more reasonable. Indeed, below we can see that in c2nse3/17.xyz, a selenium atom forms bond to the surface and induces great deformation. In c2nse3/2.xyz, the tiny selenium cluster stay in the hole of the surface, which may probably be what we expect.
