5.3. Example: Build Parameter File for (NHCH3)2CO
When your interested molecule is absent in misc/charmm36, you will have to build its parameter file by yourself. Here we will take N,N’-dimethylurea, i.e., \(\left(\mathrm{NHCH}_3\right)_2\mathrm{CO}\), as an example.
5.3.1. New Way: Automatically Building
Attention
When using topgen for this job, you are actually using some graph representation learning algorithm for automatic atom typing. Thus please cite the following paper:
Zhang, J. Atom Typing Using Graph Representation Learning: How Do Models Learn Chemistry? J. Chem. Phys. 2022, 156, 204108.
From ABCluster 3.1, the parameter file for an arbitrary organic molecule can be build automatically with topgen.
Step 1: Build the coordinate file of \(\left(\mathrm{NHCH}_3\right)_2\mathrm{CO}\) in XYZ format. Let’s call it nndimethylurea.xyz. The coordinates can be optimized as a moderate level of theory. We use B3LYPD3/def2-TZVP.
14
dimethylurea
C -3.40944156863935 1.48217854479510 0.18460221407169
O -2.83617751600667 2.55594436393475 0.29160616578062
N -4.77159876631838 1.40346427888521 0.01033470214576
H -5.17936611057651 2.32373985124644 -0.02398130498834
N -2.71942098770917 0.27746301048357 0.25258402399382
H -1.74237705636477 0.48553039432599 0.40014513551919
C -5.63042059274161 0.33540415616324 0.49938800446410
H -6.45368224181311 0.76470101295480 1.07232563209061
H -6.05781648140388 -0.26211048052894 -0.31127284718113
H -5.06868507307871 -0.32252499680997 1.16139152177693
C -2.93625253356738 -0.80249737836091 -0.70510480675735
H -2.73548051116983 -0.49573138113413 -1.73834868327471
H -2.27102803030176 -1.62710593941345 -0.45152402911762
H -3.95491690030879 -1.17972228654170 -0.65012968852358
The molecular structure and atom indices are shown below.
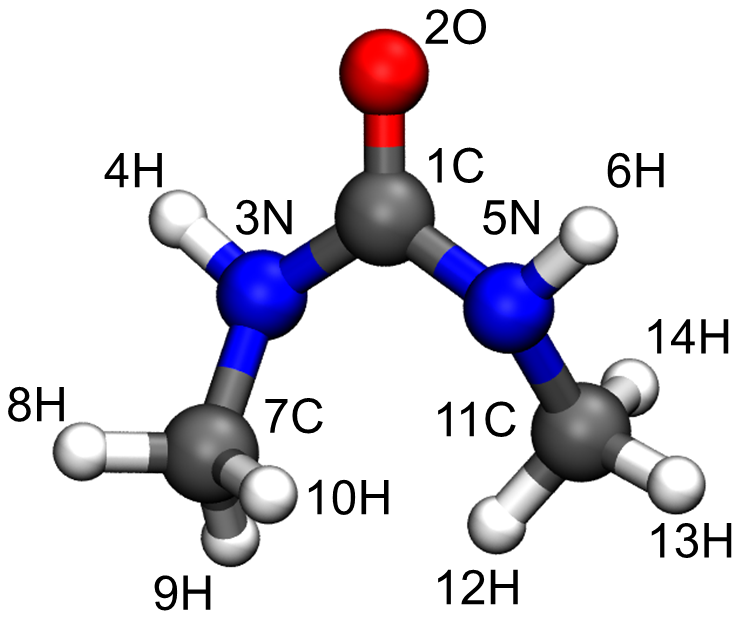
Step 2: Use topgen to build parameter files:
$ topgen nndimethylurea.xyz
Step 3: You can find the following output:
1In "nndimethylurea-rigid.xyz" and "nndimethylurea.psf":
2"X" means unknown atomic type.
3Total charge: 0.5900
So, nndimethylurea-rigid.xyz is the parameter file we need. But its total charge is 0.5900, which is not what we want. You must adjust the charges manually, or re-fit some physically meaningful ones, like RESP charges with Multiwfn. See here for a complete example.
5.3.2. Old (and out-of-date) Way: Manually Building
Step 1: Build the coordinate file of \(\left(\mathrm{NHCH}_3\right)_2\mathrm{CO}\) in XYZ format. Let’s call it nndimethylurea.xyz. The coordinates can be optimized as a moderate level of theory. We use B3LYPD3/def2-TZVP.
14
dimethylurea
C -3.40944156863935 1.48217854479510 0.18460221407169
O -2.83617751600667 2.55594436393475 0.29160616578062
N -4.77159876631838 1.40346427888521 0.01033470214576
H -5.17936611057651 2.32373985124644 -0.02398130498834
N -2.71942098770917 0.27746301048357 0.25258402399382
H -1.74237705636477 0.48553039432599 0.40014513551919
C -5.63042059274161 0.33540415616324 0.49938800446410
H -6.45368224181311 0.76470101295480 1.07232563209061
H -6.05781648140388 -0.26211048052894 -0.31127284718113
H -5.06868507307871 -0.32252499680997 1.16139152177693
C -2.93625253356738 -0.80249737836091 -0.70510480675735
H -2.73548051116983 -0.49573138113413 -1.73834868327471
H -2.27102803030176 -1.62710593941345 -0.45152402911762
H -3.95491690030879 -1.17972228654170 -0.65012968852358
The molecular structure and atom indices are shown below.
Step 2: Add a line of comment, which can be anything:
14
dimethylurea
C -3.40944156863935 1.48217854479510 0.18460221407169
O -2.83617751600667 2.55594436393475 0.29160616578062
N -4.77159876631838 1.40346427888521 0.01033470214576
H -5.17936611057651 2.32373985124644 -0.02398130498834
N -2.71942098770917 0.27746301048357 0.25258402399382
H -1.74237705636477 0.48553039432599 0.40014513551919
C -5.63042059274161 0.33540415616324 0.49938800446410
H -6.45368224181311 0.76470101295480 1.07232563209061
H -6.05781648140388 -0.26211048052894 -0.31127284718113
H -5.06868507307871 -0.32252499680997 1.16139152177693
C -2.93625253356738 -0.80249737836091 -0.70510480675735
H -2.73548051116983 -0.49573138113413 -1.73834868327471
H -2.27102803030176 -1.62710593941345 -0.45152402911762
H -3.95491690030879 -1.17972228654170 -0.65012968852358
all36_cgenff q epsilon (kJ/mol) sigma (AA)
Step 3: The force field parameters have to be derived from official CHARMM documents. They can be downloaded from here.
Tip
In CHARMM force field, each atom has a type, which is determined by its chemical environment. Most useful parameters can be found in top_all36_cgenff.rtf and par_all36_cgenff.prm.
Step 4: The paramters must be given in the cooridnate order. Now we are looking for the atom types that are similar to the ones this molecule. Let’s first dtermine atomic charges. We can find urea molecule in top_all36_cgenff.rtf:
RESI UREA 0.00 ! CH4N2O, Urea, adm
GROUP
ATOM N1 NG2S2 -0.69
ATOM H11 HGP1 0.34
ATOM H12 HGP1 0.34
ATOM C2 CG2O6 0.60
ATOM O2 OG2D1 -0.58
ATOM N3 NG2S2 -0.69
ATOM H31 HGP1 0.34
ATOM H32 HGP1 0.34
So, it is very reasonable to set atomic charges to the same value as they are in urea:
14
dimethylurea
C -3.40944156863935 1.48217854479510 0.18460221407169
O -2.83617751600667 2.55594436393475 0.29160616578062
N -4.77159876631838 1.40346427888521 0.01033470214576
H -5.17936611057651 2.32373985124644 -0.02398130498834
N -2.71942098770917 0.27746301048357 0.25258402399382
H -1.74237705636477 0.48553039432599 0.40014513551919
C -5.63042059274161 0.33540415616324 0.49938800446410
H -6.45368224181311 0.76470101295480 1.07232563209061
H -6.05781648140388 -0.26211048052894 -0.31127284718113
H -5.06868507307871 -0.32252499680997 1.16139152177693
C -2.93625253356738 -0.80249737836091 -0.70510480675735
H -2.73548051116983 -0.49573138113413 -1.73834868327471
H -2.27102803030176 -1.62710593941345 -0.45152402911762
H -3.95491690030879 -1.17972228654170 -0.65012968852358
all36_cgenff q epsilon (kJ/mol) sigma (AA)
+0.60
-0.58
-0.69
+0.34
-0.69
+0.34
Now we have to determine charges of the two methyl groups. After exmining top_all36_cgenff.rtf, we find that the charge on \(\mathrm{H}\) from \(\mathrm{CH}_3\) is almost always +0.09, then the carbon charges can be determined by requiring the total charge being zero. Now, all charges are available:
14
dimethylurea
C -3.40944156863935 1.48217854479510 0.18460221407169
O -2.83617751600667 2.55594436393475 0.29160616578062
N -4.77159876631838 1.40346427888521 0.01033470214576
H -5.17936611057651 2.32373985124644 -0.02398130498834
N -2.71942098770917 0.27746301048357 0.25258402399382
H -1.74237705636477 0.48553039432599 0.40014513551919
C -5.63042059274161 0.33540415616324 0.49938800446410
H -6.45368224181311 0.76470101295480 1.07232563209061
H -6.05781648140388 -0.26211048052894 -0.31127284718113
H -5.06868507307871 -0.32252499680997 1.16139152177693
C -2.93625253356738 -0.80249737836091 -0.70510480675735
H -2.73548051116983 -0.49573138113413 -1.73834868327471
H -2.27102803030176 -1.62710593941345 -0.45152402911762
H -3.95491690030879 -1.17972228654170 -0.65012968852358
all36_cgenff q epsilon (kJ/mol) sigma (AA)
+0.60
-0.58
-0.69
+0.34
-0.69
+0.34
+0.07
+0.09
+0.09
+0.09
+0.07
+0.09
+0.09
+0.09
Step 5: Let’s determine the Lennard-Jones parameters. It is easier: simply search the atomic type. For example, 1C in urea is of type CG2O6, then search in par_all36_cgenff.prm and you will find this:
CG2O5 0.0 -0.0900 2.00 ! adm, acetone, 11/08
CG2O6 0.0 -0.0700 2.00 ! UREA, CO3 (carbonate) from acetate heat of solvation
CG2O7 0.0 -0.0580 1.5630 ! carbon dioxide, JES
So, the two parameters are -0.0700 and 2.00. However, they must be transformed before using in ABCluster. The formulas are:
\(\epsilon_{\mathrm{ABCluster}} = \epsilon_{\mathrm{CHARMM}}\times(-4.184)\) |
\(\sigma_{\mathrm{ABCluster}} = \sigma_{\mathrm{CHARMM}}\times 2^{5/6}\) |
Therefore:
We can put these parameters to nndimethylurea.xyz:
14
dimethylurea
C -3.40944156863935 1.48217854479510 0.18460221407169
O -2.83617751600667 2.55594436393475 0.29160616578062
N -4.77159876631838 1.40346427888521 0.01033470214576
H -5.17936611057651 2.32373985124644 -0.02398130498834
N -2.71942098770917 0.27746301048357 0.25258402399382
H -1.74237705636477 0.48553039432599 0.40014513551919
C -5.63042059274161 0.33540415616324 0.49938800446410
H -6.45368224181311 0.76470101295480 1.07232563209061
H -6.05781648140388 -0.26211048052894 -0.31127284718113
H -5.06868507307871 -0.32252499680997 1.16139152177693
C -2.93625253356738 -0.80249737836091 -0.70510480675735
H -2.73548051116983 -0.49573138113413 -1.73834868327471
H -2.27102803030176 -1.62710593941345 -0.45152402911762
H -3.95491690030879 -1.17972228654170 -0.65012968852358
all36_cgenff q epsilon (kJ/mol) sigma (AA)
+0.60 0.2929 3.5636 # CG2O6
-0.58
-0.69
+0.34
-0.69
+0.34
+0.07
+0.09
+0.09
+0.09
+0.07
+0.09
+0.09
+0.09
Note that we can add a comment at the end of parameters.
Step 6: In a similar way, we can build all parameters for this molecule:
14
dimethylurea
C -3.40944156863935 1.48217854479510 0.18460221407169
O -2.83617751600667 2.55594436393475 0.29160616578062
N -4.77159876631838 1.40346427888521 0.01033470214576
H -5.17936611057651 2.32373985124644 -0.02398130498834
N -2.71942098770917 0.27746301048357 0.25258402399382
H -1.74237705636477 0.48553039432599 0.40014513551919
C -5.63042059274161 0.33540415616324 0.49938800446410
H -6.45368224181311 0.76470101295480 1.07232563209061
H -6.05781648140388 -0.26211048052894 -0.31127284718113
H -5.06868507307871 -0.32252499680997 1.16139152177693
C -2.93625253356738 -0.80249737836091 -0.70510480675735
H -2.73548051116983 -0.49573138113413 -1.73834868327471
H -2.27102803030176 -1.62710593941345 -0.45152402911762
H -3.95491690030879 -1.17972228654170 -0.65012968852358
all36_cgenff q epsilon (kJ/mol) sigma (AA)
+0.60 0.2929 3.5636 # CG2O6
-0.58 0.5021 3.0291 # OG2D1
-0.69 0.8368 3.2963 # NG2S2
+0.34 0.1925 0.4000 # HGP1
-0.69 0.8368 3.2963 # NG2S2
+0.34 0.1925 0.4000 # HGP1
+0.07 0.3264 3.6527 # CG331
+0.09 0.1004 2.3876 # HGA3
+0.09 0.1004 2.3876 # HGA3
+0.09 0.1004 2.3876 # HGA3
+0.07 0.3264 3.6527 # CG331
+0.09 0.1004 2.3876 # HGA3
+0.09 0.1004 2.3876 # HGA3
+0.09 0.1004 2.3876 # HGA3
Now we can use this nndimethylurea.xyz for global optimization!
Tip
When build your own parameter files, note that the charges can be slightly adjusted according to the total charge or chemical environments, but the Lennard-Jones parameters should be kept as they are!
Before conclusion, we point out that rigidmol supports virtual sites. For example, in TIP4P water, a virtual site exists close to the oxygen atom. The symbol of all virtual sites starts with q. For example, in misc/charmm36/tip4p.xyz:
4
H2O
O 0.00000000 0.00000000 0.21451132
qLP 0.00000000 0.00000000 0.06451132
H 0.00000000 0.75695033 -0.37137096
H 0.00000000 -0.75695033 -0.37137096
TIP4P q epsilon (kJ/mol) sigma (AA)
0.000 0.64852 3.1536 # OT
-1.040 0.00000 0.0000 # LP
+0.520 0.00000 0.0000 # HT
+0.520 0.00000 0.0000 # HT
qLP is the virtual site. Note that in output files, all q* will be written in *.xyz but not in *.gjf. You can use this feature to design some special force fields.