Tip
All input files can be downloaded: Files.
eda
This keyword defines how to perform an energy decomposition analysis (EDA) calculation.
Options
- type
Value
tsofor block localized wavefunction energy decomposition analysis (TSO-EDA).gksfor generalized Kohn-Sham energy decomposition analysis (GKS-EDA).mb_tsofor many-body interaction TSO-EDA.mb_gksfor many-body interaction GKS-EDA.Default
None
Both GKS-EDA and TSO-EDA is available for intermolecular interaction analysis. Furthermore, combined with the many-body expansion scheme, two new methods, i.e. many-body GKS-EDA and many-body TSO-EDA, are available for the analysis of many-body interactions.
In Qbics, we recommend to use TSO-EDA for EDA calculations.
- frag
This option defines the fragments’ partition of an system. The format is:
frag num_electrons spin_multiplicity atom_rangewhich is the same as the keyword
fraginscfguessoption. See scfguess.
- nobsse
Do not do the Boys and Bernardi’s counterposise (CP) correction for basis set superposition error (BSSE). Qbics does BSSE correction by default. You can use this keword to avoid it when you don’t need to consider BSSE.
- tso_for_guess
Do TSO calculation first for the initial guess of fragments’ wavefunction, which is necessary for the case there are fragments with C∞ group symmetry such as an atom.
For
tso`andmb_tso, this is default. While forgksandmb_gks, this is optional.
- mb_level
Value
An integer
Default
2Truncation level for many-body interaction analysis, i.e.
mb_gksandmb_tsocalculations. The value should NOT be smaller than 2 and equal to or greater than the number of fragments.Usually,
4is a good choice, if the number of fragments is larger than 4. Higher order terms are very small and can be ignored.
Warning
For EDA tasks, you should add type U in the scf keyword to ensure the unrestricted calculation. Otherwish, the calculation will be failed.
Theoretical Background
Hint
If you use tso and mb_tso, please cite the following reference:
If you use gks and mb_gks, please cite the following reference:
TSO-EDA
TSO-EDA is based on target state optimization self-consistent field method (J. Chem. Theory Comput. 2023, 19, 1777) and decomposes the total interaction energy into five terms, i.e. electrostatic, exchange, polarization, charge transfer, and dispersion energies. The sum of electrostattic and exchange energy is the Heitler-London term (Phys. Chem. Chem. Phys. 2024, 26, 17549):
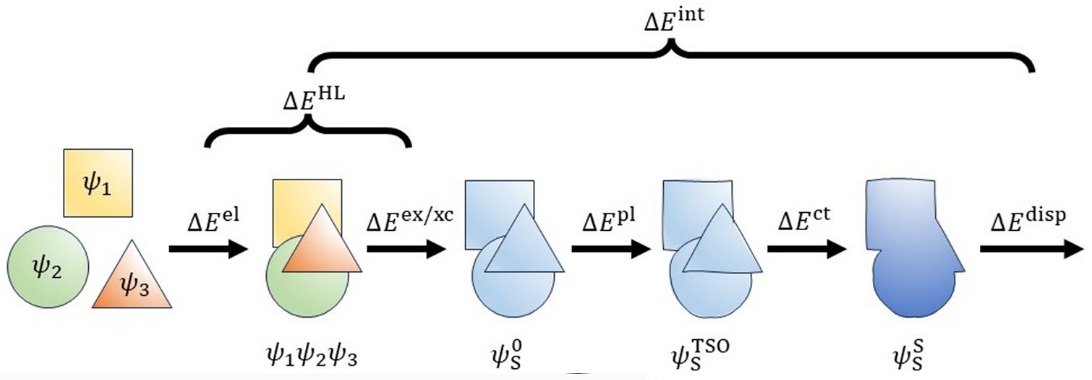
Here:
Ectrostatic term: Represents the semiclassical Coulombic interaction of charged particles from different monomers;
Exchange term: Represents quantum effect due to the antisymmetric character of the electronic wave function and the satisfaction of the Pauli exclusion principle;
Polarization term: Represents the polarization of the electron density of one monomer by the presence of other monomer;
Charge transfer term: Represents the charge transfer between monomers;
Dispersion term: Represents the dispersion interaction between monomers.
The BSSE effect is included in charge transfer term. All above terms can be found in Qbics output.
Many-Body TSO-EDA
This method is developed in Phys. Chem. Chem. Phys. 2024, 26, 17549 and is used to analyze the many-body effects in a molecular cluster. The total interaction energy is decomposed into 2-body, 3-body, and higher-order terms, like this:
The terms higher than \(\Delta E^{(2)}\) is the many-body interaction term. Usually the most important one is the three-body effect \(\Delta E^{(3)}\), the effects of which can be decomposed into three ones:
\(\Delta E^{(3)} < 0\): Indicate a cooperative effect of the monomers in a cluster. The many-body interaction is stabilizing the cluster. This is often seen in hydrogen bonding clusters, like water clusters.
\(\Delta E^{(3)} > 0\): Indicate an anti-cooperative effect of the monomers in a cluster. The many-body interaction is destabilizing the cluster. This is often seen in a cluster of charged species, like ionic liquid clusters. Also see below.
\(\Delta E^{(3)} \approx 0\): Indicate a non-cooperative effect of the monomers in a cluster. There is little many-body interaction in the cluster. This is often seen in a cluster of molecules without charges or hydrogen bonds.
Each order can be decomposed into electrostatic, exchange, polarization, charge transfer, and dispersion terms:
Usually, electrostatic and exchange terms are highly additive, while polarization and charge transfer terms are non-additive. The dispersion term is always additive.
Input Examples
Example: EDA for GeH3F-NCH Complex by B3LYP-D3BJ/def2-SVP
For the complex GeH3F-NCH, we can do EDA calculation by the following input:
1mol
2 Ge 0.00000000 0.00221863 -0.79935317
3 H 0.00000000 1.48645043 -0.40384625
4 H 1.28514604 -0.74161126 -0.40477816
5 H -1.28514603 -0.74161126 -0.40477816
6 F 0.00000000 0.00108752 -2.56116087
7 C 0.00000000 -0.00225138 3.35662076
8 H 0.00000000 -0.00220444 4.43604901
9 N 0.00000000 -0.00207825 2.20326200
10end
11
12basis
13 def2-svp
14end
15
16scf
17 charge 0
18 spin2p1 1
19 type U # For EDA calculations, this must be added explicitly.
20end
21
22grimmedisp
23 type bj
24end
25
26eda
27 type tso # You can also change it to: gks
28 frag 0 1 1-5 # Define GeH3F.
29 frag 0 1 6-8 # Define HCN.
30end
31
32task
33 eda b3lyp
34end
The atom indices are shown below:
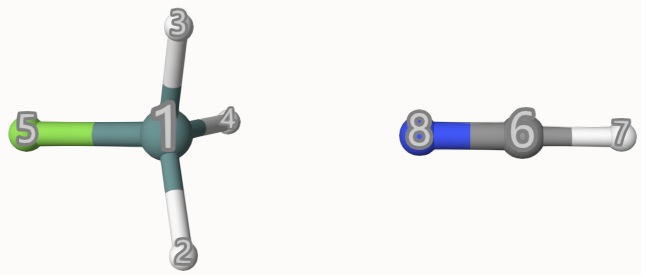
The results are:
1WITH BSSE correction:
2Electrostatic interaction energy: -4.98 kcal/mol
3Exchange-correlation interaction energy: 4.22 kcal/mol
4Polarization interaction energy: -0.62 kcal/mol
5Charge transfer interaction energy: -1.31 kcal/mol
6Grimme's dispersion interaction: -1.58 kcal/mol
7----------------------------------------------------------------
8Total interaction energy: -4.27 kcal/mol
1WITH BSSE correction:
2Electrostatic interaction energy: -6.22 kcal/mol
3Exchange interaction energy: -9.64 kcal/mol
4Repulsion interaction energy: 15.94 kcal/mol
5Polarization interaction energy: -2.52 kcal/mol
6Correlation interaction energy: -0.24 kcal/mol
7Grimme's dispersion interaction: -1.58 kcal/mol
8----------------------------------------------------------------
9Total interaction energy: -4.27 kcal/mol
We can see that the total interaction energies (with or without BSSE) are the same for both TSO-EDA and GKS-EDA methods, but components are different. As mentioned, Qbics recommends TSO-EDA for calculations. This complex is stabilized by Electrostatic interaction eneregy, which is compatible with the chemical intuition that it is stabilized by sigma-hole.
Example: MB-EDA for Molecular Cluster (NH4+)2(H2SO4)(HSO4-)2
The title cluster is composed of two NH4+ cations, one H2SO4 molecules, and two HSO4- anions. This cluster is used in the study of atmopheric chemistry.We can do MB-EDA calculation by the following input:
1basis
2 def2-svp
3end
4
5scf
6 charge 0 # Total charge.
7 spin2p1 1
8 type U
9end
10
11grimmedisp
12 type bj
13end
14
15eda
16 type mb_tso
17 mb_level 4
18 frag +1 1 1-5 # NH4+
19 frag +1 1 6-10 # NH4+
20 frag 0 1 11-17 # H2SO4
21 frag -1 1 18-23 # HSO4-
22 frag -1 1 24-29 # HSO4-
23end
24
25mol
26 N 0.13124700 -1.86033100 -1.49054300
27 H -0.68471400 -1.96085700 -0.84840100
28 H 0.16284500 -2.63375000 -2.14527600
29 H -0.00155300 -0.97157900 -1.98611500
30 H 1.02982000 -1.79400200 -0.97437700
31 N -1.89606400 2.02266900 1.95536400
32 H -2.33766600 1.07911300 1.78190600
33 H -1.20423600 1.92734800 2.69193100
34 H -1.40455300 2.34660500 1.08417600
35 H -2.60508400 2.69280200 2.23215700
36 S 3.40269500 -0.73966700 0.43845300
37 O 4.56636300 -1.26003200 1.03924300
38 O 2.66268100 -1.55477200 -0.49575900
39 O 2.42657400 -0.30120000 1.56959800
40 O 3.78755300 0.58843400 -0.27018200
41 H 2.99297400 1.01172300 -0.68798600
42 H 1.56137200 -0.00498400 1.17228000
43 S -3.05756300 -0.82805000 0.17173500
44 O -2.21824200 -1.98280100 -0.09354400
45 O -3.00471800 -0.39464500 1.56194000
46 O -2.90973700 0.26502300 -0.77053900
47 O -4.55472700 -1.30387800 -0.08712900
48 H -4.73898300 -2.05912100 0.48328300
49 H -1.51856700 0.72871100 -1.53329100
50 S 0.24159900 1.52238500 -0.65825900
51 O -0.59336700 0.90131200 -1.85962900
52 O 1.55183900 1.72430300 -1.22252400
53 O -0.45708500 2.72297400 -0.23978600
54 O 0.20716100 0.49864900 0.39894800
55end
56
57task
58 eda b3lyp
59end
The atom indices are shown below, which is used to define the fragments frag:
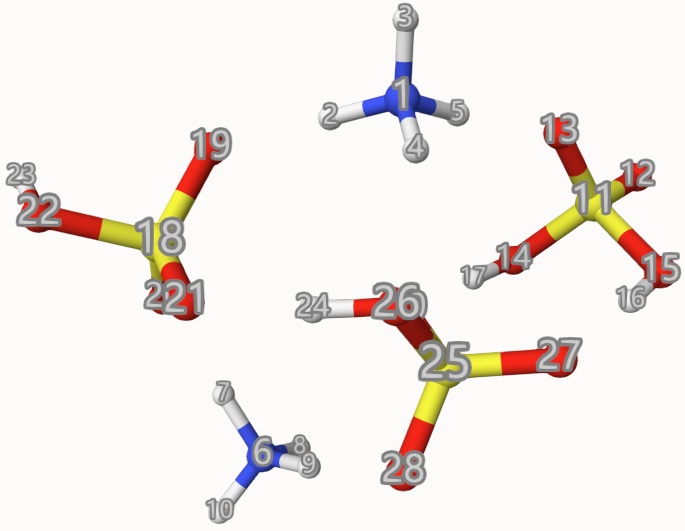
The results are:
1Table 5. Summary (kcal/mol).
2---------------------------------------------------------------------------------------------------------------------------------
3 Interactions delE_el delE_xc delE_pl delE_ct delE_bsse delE_disp delE_tot
4---------------------------------------------------------------------------------------------------------------------------------
5 SUM of 2-body -3.51853640E+02 1.04231044E+02 -5.50310217E+01 -9.70290703E+01 4.35696096E+01 -1.98079059E+01 -3.75920984E+02
6 SUM of 3-body 1.72107484E-09 1.45358519E+00 2.82254671E+01 2.81735373E+01 -1.24450163E+01 1.01531078E-02 4.54177264E+01
7 SUM of 4-body -4.14670076E-09 1.70212282E-02 -2.25462767E+00 -5.15894065E+00 1.89758583E+00 2.28017787E-05 -5.49893846E+00
8 Remain 7.51748885E-09 -8.58522126E-04 6.43113307E-02 4.30266615E-01 -1.22896862E-01 5.97720460E-07 3.70823167E-01
9---------------------------------------------------------------------------------------------------------------------------------
10 SUM -3.51853640E+02 1.05700792E+02 -2.89958710E+01 -7.35842070E+01 3.28992822E+01 -1.97977294E+01 -3.35631373E+02
11---------------------------------------------------------------------------------------------------------------------------------
We can see that the total interaction energy is -335.63 kcal/mol, which is decomposed into 2-body, 3-body, 4-body, and remaining terms. The 2-body term is the most important one (-375.92 kcal/mol), while the 3-body term is also significant, but anti-cooperative (destablizing the complex) (+45.42 kcal/mol). The 4-body term is small (-5.50 kcal/mol, slightly cooperative). The remaining term (sum of 5- and 6-body) is very small (+0.37 kcal/mol) and can be ignored.
We can also see that the electrostatic and exchange energy are highly additive, while the polarization and charge-transfer energy are non-additive. For different kinds of clusters, the 3-body effects (many-body interactions) can be quite different, see Phys. Chem. Chem. Phys. 2024, 26, 17549 for more information.