Tip
All input files can be downloaded: Files.
Tip
Please refer to scfguess for more examples.
For a complete tutorial of TSO-DFT, please refer to:
scf
This option controls how to perform an SCF calculation.
Options
- charge
Value
An integer
Default
0Define the total charge of the system.
- spin2p1
Value
An integer
Default
1for even number of electrons2for odd number of electronsDefine the spin multiplicity of the system, i.e., \(2S+1\), where \(S\) is the spin of the system. For example, to consider the singlet and triplet state, one should set
spin2p1to1and3, respectively.Note that
spin2p1can be either positive or negative. Both positive and negativespin2p1represent the same spin multiplicity, but for a SCF wave function, alpha and beta orbitals will be occupied first, respectively. For example, for 11 electrons withspin2p1being1, there will be 6 alpha electrons and 5 beta electrons, like most quantum chemistry software does; but whenspin2p1is-1, there will be 5 alpha electrons and 6 beta electrons.For odd number of electrons, a positive
spin2p1will first occupy alpha orbitals. If you want the beta orbitals to be occupied first, use a negativespin2p1. For example:1scf 2 charge 0 3 spin2p1 3 4end 5mol 6 H 0. 0. 0. 7 H 0. 0. 0.74 8end
In this case, the molecule will have 2 alpha electrons and 0 beta ones. For another input:
1scf 2 charge 0 3 spin2p1 -3 4end 5mol 6 H 0. 0. 0. 7 H 0. 0. 1. 8end
The molecule will have 2 beta electrons and 0 alpha ones.
- type
Value
Rfor restricted SCF (alpha and beta orbitals are restricted to be identical)Ufor unrestricted SCF (alpha and beta orbitals are not necessarily identical)Default
Rfor singlet stateUfor other statesFor singlet states, both restricted and unrestricted SCF are available. Unrestricted SCF is very useful in treating spin polarized systems. For non-singlet states, only the unrestricted one can be used.
- max_it
Value
A non-negative integer
Default
128Define the maximum number of SCF iteration.
Hint
You can set
max_itto0to do a non-iterative calculation.
- energy_cov
Value
A real number
Default
1.E-6The energy convergence threshold for SCF calculations.
- density_cov
Value
A real number
Default
1.E-8The density matrix convergence threshold for SCF calculations.
Hint
The SCF calculation is determined to be convergent when both the energy and density convergence conditions are satisfied.
Hint
If SCF does not converge, Qbics will exit immediately. If you want Qbics to continue even an SCF calculation does not converge, please refer to output.
- no_diis
Do not use direct inversion of the iterative subspace (DIIS) convergence acceleration algorithm.
- no_damping
Do not use damping convergence acceleration algorithm.
- damping_factor
Value
A real number
Default
0.3The density matrix damping factor to accelerate SCF convergence.
- print_MO
Print molecular orbital coefficients. Without this, only molecular orbital energies and occupancies are printed.
- schwarz
Value
A real number
Default
1.E-10Define the Schwarz screening tolerance. All two-electron integral contributions below this tolerance will be discarded to speed up calculations. A positive real number is needed.
Warning
Do not set a too large value (like
1.E-5). It may leads to wrong results.
- no_scf
Value
tsoDo target state optimization SCF (TSO-SCF).bleDo block-localized excitation SCF (BLE-SCF).Default
None
Instead of doing ordinary SCF, do non-orthogonal SCF to treat diabatic and excited states. This is a powerful method. Some input examples can be found in scfguess. For theoretical details, please refer to:
Tip
For a complete tutorial of TSO-DFT, please refer to:
Theoretical Background
The self-consistent field (SCF) method is the corner stone of modern electronic theory and computational chemistry. In Qbics, both Hartree-Fock and Kohn-Sham methods can be used. After calculations, energies are given in the output file, and the wave function is given in an MWFN file, which can be visualzied in Qbics-MolStar or Multiwfn.
Density Functionals
For Kohn-Sham methods, the available density functionals are listed below:
Local density approximation (LDA)
ldaGeneralized gradient approximation (GGA)
pbe
blyp
bp86
olyp
pw91Hybrid GGA
pbe0
b3lyp
o3lyp
b3pw91
x3lypMeta-GGA
tpss
m06lHybrid meta-GGA
tpssh
m06
m062x
m06hf
In modern computational chemistry, there have been a lot of experience for basis functional selection. Here, we just give a few points:
In most cases, LDA should NOT be used.
For organic compounds, hybrid functionals B3LYP and M06-2X are often the most reliable ones.
For compounds containing metals, GGAs like BP86 and BLYP sometimes work better than hybrid GGAs.
Of course, the best way for basis functional selection is to do calibration for your specific system.
Also, in all modern calculations, one should use disperison corrections, like DFT-D3 in grimmedisp.
SCF Convergence
In Qbics, if an SCF does not converg, please try the following methods:
Increase number of iterations, like
mat_it 200, although in most cases this does not work;Set
schwarzto a small value, likeschwarz 1E-14;Use a converged wave function as the initial guess, say the same system with smaller basis sets. See scfguess for details.
Non-orthogonal SCF: TSO-DFT
XXXXXXXXXXXXXX
Non-orthogonal SCF: BLE-DFT
In QBics, the BLE (Block-Localized Excitation) method is implemented using the TSO (Two-Step Optimization) module to divide orbitals into blocks. In each block, a block-localized SCF equation is solved.
The only difference between BLE and standard TSO is in the treatment of excited-state configurations:
BLE combines the block-SCF framework with the Δ-SCF method proposed by Peter Gill et al., rather than simply excluding certain orbitals.
Initial Maximum Overlap Method (IMOM) is employed:
After the reference calculation, the excitation configuration is defined by
scfguess ble.The program stores the occupied orbital coefficients as \(\mathbf{C}^{\text{old}}_{\text{occ}}\).
These are then used to construct a new Fock matrix, which is diagonalized to obtain new coefficients \(\mathbf{C}^{\text{new}}\).
Instead of assigning occupations based on orbital energies, BLE uses the overlap projection between new and old occupied orbitals.
The projection is evaluated by:
Orbitals with the largest projections are selected as the newly occupied orbitals.
Input Examples
Note that, in all examples below, you can change energy to opt to do geometry optimization, or md to do molecular dynamics.
Example: DFT Energy of Dieldrin
In scf-1a.inp, scf-1b.inp, and scf-1c.inp, we calculate the singlet state, triplet state, and cationic state of dieldrin. For cationic state of dieldrin, since an electron is lost, its spin is 2 so it is a doublet state. There are controled by charge and spin2p1.
1basis
2 def2-svp
3end
4
5scf
6 charge 0
7 spin2p1 1
8end
9
10grimmedisp
11 type bj
12end
13
14mol
15 Cl 1.40900 -0.54900 -0.14200
16 C 2.91800 0.22200 -0.14800
17 C 3.34900 1.18000 -0.99000
18 Cl 2.48900 1.91800 -2.24800
19 C 4.73000 1.60600 -0.53700
20 Cl 5.24600 3.22500 -0.97500
21 C 5.80600 0.51900 -0.74700
22 C 6.83600 -0.13400 -1.70000
23 C 6.40300 -1.62200 -1.82900
24 C 6.19900 -1.81300 -0.26300
25 C 7.62200 -1.50500 0.16600
26 O 8.02000 -0.22000 0.59300
27 C 8.09500 -0.42900 -0.82100
28 C 5.11900 -0.73000 -0.15200
29 C 4.07300 -0.11600 0.82100
30 Cl 3.68500 -1.10500 2.20000
31 C 4.50000 1.37200 1.05000
32 Cl 5.90100 1.54700 2.17100
33 Cl 3.22100 2.43400 1.80000
34 H 6.52400 1.00200 -0.09500
35 H 7.05500 0.38900 -2.63100
36 H 7.19900 -2.27000 -2.21800
37 H 5.50600 -1.77200 -2.43800
38 H 5.86900 -2.82100 -0.00200
39 H 8.25600 -2.36000 0.34800
40 H 9.03900 -0.55400 -1.32700
41 H 4.42500 -1.17200 -0.89200
42end
43
44task
45 energy b3lyp
46end
1basis
2 def2-svp
3end
4
5scf
6 charge 0
7 spin2p1 3
8end
9
10grimmedisp
11 type bj
12end
13
14mol
15 Cl 1.40900 -0.54900 -0.14200
16 C 2.91800 0.22200 -0.14800
17 C 3.34900 1.18000 -0.99000
18 Cl 2.48900 1.91800 -2.24800
19 C 4.73000 1.60600 -0.53700
20 Cl 5.24600 3.22500 -0.97500
21 C 5.80600 0.51900 -0.74700
22 C 6.83600 -0.13400 -1.70000
23 C 6.40300 -1.62200 -1.82900
24 C 6.19900 -1.81300 -0.26300
25 C 7.62200 -1.50500 0.16600
26 O 8.02000 -0.22000 0.59300
27 C 8.09500 -0.42900 -0.82100
28 C 5.11900 -0.73000 -0.15200
29 C 4.07300 -0.11600 0.82100
30 Cl 3.68500 -1.10500 2.20000
31 C 4.50000 1.37200 1.05000
32 Cl 5.90100 1.54700 2.17100
33 Cl 3.22100 2.43400 1.80000
34 H 6.52400 1.00200 -0.09500
35 H 7.05500 0.38900 -2.63100
36 H 7.19900 -2.27000 -2.21800
37 H 5.50600 -1.77200 -2.43800
38 H 5.86900 -2.82100 -0.00200
39 H 8.25600 -2.36000 0.34800
40 H 9.03900 -0.55400 -1.32700
41 H 4.42500 -1.17200 -0.89200
42end
43
44task
45 energy b3lyp
46end
1basis
2 def2-svp
3end
4
5scf
6 charge +1
7 spin2p1 2
8end
9
10grimmedisp
11 type bj
12end
13
14mol
15 Cl 1.40900 -0.54900 -0.14200
16 C 2.91800 0.22200 -0.14800
17 C 3.34900 1.18000 -0.99000
18 Cl 2.48900 1.91800 -2.24800
19 C 4.73000 1.60600 -0.53700
20 Cl 5.24600 3.22500 -0.97500
21 C 5.80600 0.51900 -0.74700
22 C 6.83600 -0.13400 -1.70000
23 C 6.40300 -1.62200 -1.82900
24 C 6.19900 -1.81300 -0.26300
25 C 7.62200 -1.50500 0.16600
26 O 8.02000 -0.22000 0.59300
27 C 8.09500 -0.42900 -0.82100
28 C 5.11900 -0.73000 -0.15200
29 C 4.07300 -0.11600 0.82100
30 Cl 3.68500 -1.10500 2.20000
31 C 4.50000 1.37200 1.05000
32 Cl 5.90100 1.54700 2.17100
33 Cl 3.22100 2.43400 1.80000
34 H 6.52400 1.00200 -0.09500
35 H 7.05500 0.38900 -2.63100
36 H 7.19900 -2.27000 -2.21800
37 H 5.50600 -1.77200 -2.43800
38 H 5.86900 -2.82100 -0.00200
39 H 8.25600 -2.36000 0.34800
40 H 9.03900 -0.55400 -1.32700
41 H 4.42500 -1.17200 -0.89200
42end
43
44task
45 energy b3lyp
46end
In the output, we can find energies:
1Mulliken Populations
2====================
3 # Symbol Charge Spin
4----------------------------------------------
5 1 Cl -0.02667453 0.00000000
6 2 C 0.00442623 0.00000000
7 3 C 0.07960288 0.00000000
8 4 Cl -0.03329205 0.00000000
9 5 C -0.08518096 0.00000000
10 6 Cl -0.05476695 0.00000000
11 7 C 0.16951697 0.00000000
12 8 C -0.10715159 0.00000000
13 9 C 0.10427346 0.00000000
14 10 C -0.08217275 0.00000000
15 11 C 0.17187228 0.00000000
16 12 O -0.26857284 0.00000000
17 13 C 0.10869646 0.00000000
18 14 C 0.11543761 0.00000000
19 15 C 0.00080890 0.00000000
20 16 Cl -0.05286360 0.00000000
21 17 C -0.10222137 0.00000000
22 18 Cl -0.01739246 0.00000000
23 19 Cl -0.03082423 0.00000000
24 20 H 0.05649853 0.00000000
25 21 H -0.00112520 0.00000000
26 22 H 0.00490582 0.00000000
27 23 H 0.00613081 0.00000000
28 24 H -0.00349217 0.00000000
29 25 H 0.00514424 0.00000000
30 26 H 0.01085963 0.00000000
31 27 H 0.02755688 0.00000000
32----------------------------------------------
33 Sum 0.00000000 0.00000000
34----------------------------------------------
35
36Electric Multipole Moments
37==========================
38 # Total Electronic Nuclear Unit
39------------------------------------------------------------------------------------
40Charge:
41 0 -0.0000 -190.0000 190.0000 |e|
42Dipole moment:
43 X 1.5427 -4249.7734 4251.3161 Debye
44 Y -3.0998 -516.9081 513.8084 Debye
45 Z -2.9629 -61.9991 59.0362 Debye
46 Total 4.5571 Debye
47Quadrupole moment:
48 XX -131.6546 -23335.2658 23203.6113 Debye*Angstrom
49 XY -18.9204 -1824.1358 1805.2154 Debye*Angstrom
50 XZ -19.3935 -226.0675 206.6740 Debye*Angstrom
51 YY -148.1762 -2656.8691 2508.6930 Debye*Angstrom
52 YZ 0.3190 50.0250 -49.7060 Debye*Angstrom
53 ZZ -149.5476 -2104.9692 1955.4216 Debye*Angstrom
54------------------------------------------------------------------------------------
55
56 ---- Self Consistent Field Energy Done ------------------
57
58Final total energy: -3297.20331257 Hartree
1Mulliken Populations
2====================
3 # Symbol Charge Spin
4----------------------------------------------
5 1 Cl 0.00050242 0.06480421
6 2 C -0.02297255 0.42027475
7 3 C 0.04197140 0.41450595
8 4 Cl 0.00210632 0.06884730
9 5 C -0.07824196 -0.03692263
10 6 Cl -0.06050498 0.00369721
11 7 C 0.17682446 0.02328426
12 8 C -0.10859383 -0.00191029
13 9 C 0.10443565 0.00083317
14 10 C -0.08144729 0.00466281
15 11 C 0.17219425 0.00079567
16 12 O -0.26819454 0.00027313
17 13 C 0.10880086 0.00191331
18 14 C 0.11355660 0.01984239
19 15 C 0.00237412 -0.03673057
20 16 Cl -0.05782701 0.00045010
21 17 C -0.10605427 0.03134455
22 18 Cl -0.01706027 0.00709001
23 19 Cl -0.02178593 0.01323355
24 20 H 0.05573434 0.00106943
25 21 H -0.00089272 0.00053599
26 22 H 0.00453855 0.00011638
27 23 H 0.00534689 0.00001025
28 24 H -0.00360374 -0.00005744
29 25 H 0.00499224 0.00021128
30 26 H 0.01069384 0.00004035
31 27 H 0.02310712 -0.00221511
32----------------------------------------------
33 Sum -0.00000000 1.00000000
34----------------------------------------------
35
36Electric Multipole Moments
37==========================
38 # Total Electronic Nuclear Unit
39------------------------------------------------------------------------------------
40Charge:
41 0 -0.0000 -190.0000 190.0000 |e|
42Dipole moment:
43 X 1.2321 -4250.0840 4251.3161 Debye
44 Y -3.0209 -516.8293 513.8084 Debye
45 Z -3.0993 -62.1355 59.0362 Debye
46 Total 4.5000 Debye
47Quadrupole moment:
48 XX -133.5686 -23337.1799 23203.6113 Debye*Angstrom
49 XY -19.1346 -1824.3499 1805.2154 Debye*Angstrom
50 XZ -19.5174 -226.1914 206.6740 Debye*Angstrom
51 YY -148.0570 -2656.7500 2508.6930 Debye*Angstrom
52 YZ 0.5035 50.2094 -49.7060 Debye*Angstrom
53 ZZ -149.1564 -2104.5779 1955.4216 Debye*Angstrom
54------------------------------------------------------------------------------------
55
56 ---- Self Consistent Field Energy Done ------------------
57
58Final total energy: -3297.07378259 Hartree
1Mulliken Populations
2====================
3 # Symbol Charge Spin
4----------------------------------------------
5 1 Cl 0.15119087 0.05849971
6 2 C 0.02320835 0.10413699
7 3 C 0.11530715 0.10227274
8 4 Cl 0.14762665 0.06201522
9 5 C -0.10261555 -0.00892321
10 6 Cl 0.03049491 0.00815814
11 7 C 0.16533293 0.02207972
12 8 C -0.10602506 -0.00233488
13 9 C 0.09403621 0.00343824
14 10 C -0.08414937 0.00100276
15 11 C 0.18370650 0.02469158
16 12 O -0.24712743 0.01483155
17 13 C 0.12028486 0.02898236
18 14 C 0.08667493 0.01029275
19 15 C 0.00527258 -0.00828766
20 16 Cl 0.02762102 0.00448934
21 17 C -0.13876750 0.00141647
22 18 Cl 0.08563728 0.03050623
23 19 Cl 0.07042179 0.04061396
24 20 H 0.07885872 0.00024694
25 21 H 0.03698109 0.00296145
26 22 H 0.04565520 -0.00004484
27 23 H 0.02804737 0.00091680
28 24 H 0.03460665 0.00193844
29 25 H 0.05000090 -0.00137480
30 26 H 0.05564188 -0.00154950
31 27 H 0.04207709 -0.00097648
32----------------------------------------------
33 Sum 1.00000000 0.50000000
34----------------------------------------------
35
36Electric Multipole Moments
37==========================
38 # Total Electronic Nuclear Unit
39------------------------------------------------------------------------------------
40Charge:
41 0 1.0000 -189.0000 190.0000 |e|
42Dipole moment:
43 X 21.9659 -4229.3502 4251.3161 Debye
44 Y -0.5347 -514.3431 513.8084 Debye
45 Z -4.0803 -63.1165 59.0362 Debye
46 Total 22.3481 Debye
47Quadrupole moment:
48 XX -14.7654 -23218.3767 23203.6113 Debye*Angstrom
49 XY -12.5519 -1817.7672 1805.2154 Debye*Angstrom
50 XZ -22.8508 -229.5249 206.6740 Debye*Angstrom
51 YY -129.2800 -2637.9729 2508.6930 Debye*Angstrom
52 YZ -0.6554 49.0506 -49.7060 Debye*Angstrom
53 ZZ -132.5362 -2087.9578 1955.4216 Debye*Angstrom
54------------------------------------------------------------------------------------
55
56 ---- Self Consistent Field Energy Done ------------------
57
58Final total energy: -3296.89315774 Hartree
In the output files, we can find energies and propertiels like Mulliken charges and spins and electric multipole moments.
We can calculate some energies:
Triple-singlet gap:
-3297.07378259--3297.20331257= 3.52 eV;Vertical electron detachment energy:
-3296.89315774--3297.20331257= 8.44 eV;
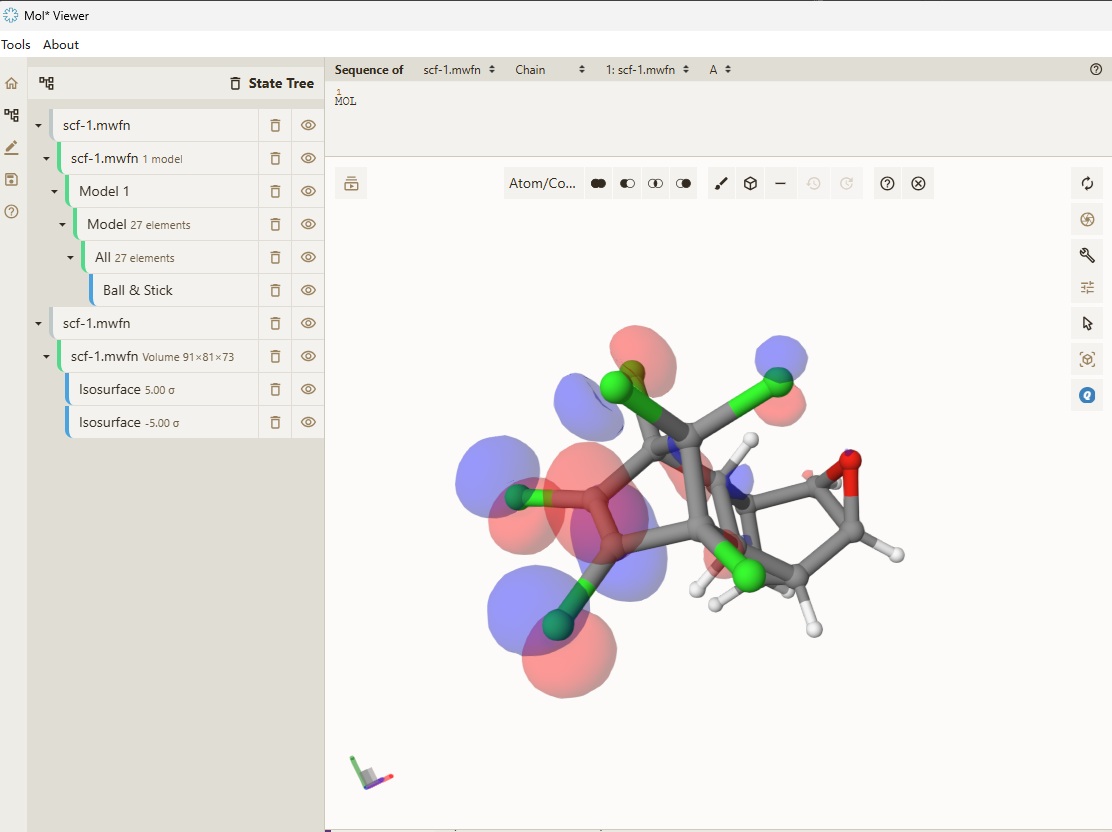
We can also visualize the molecular orbitals. Open Qbics-MolStar, and drag scf-1a.mwfn into explorer, and it will be automatically loaded. Right-click scf-1a.mwfn and select View Molecular Orbitals, 95: -0.24732 (occ=2, RHF), then the HOMO of singlet state of dieldrin is visualized:
Other wavefunction proterties can be visualized. For example, dragging scf-1b.mwfn into Qbics-MolStar, and right-click scf-1b.mwfn, then click View Other Wavefunction Properties, in Format, click Electron Spin Density, you can see spin density of this system:
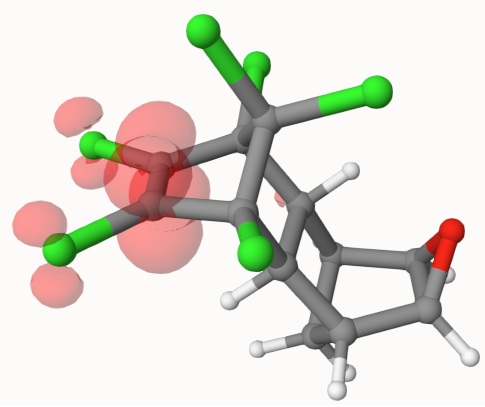
These works are supported by Multiwfn in the backend. Please cite Multiwfn accoring to http://sobereva.com/multiwfn if you use these data.
Example: TSO-DFT and BLE-DFT
For a detailed tutorial, please refer to: