Tip
All input files can be downloaded: Files.
Transition State Search
This tutorial will lead you step by step to search transition states using Qbics.
There are two kinds of methods to search transition states in Qbics:
The QST2/dimer method and the nudged elastic band (NEB) method. For both methods, a reactant and a product geometry have to be provided.
The diabatic minimum energy crossing point (dMECP) method. This method is highly useful for AB+C=A+BC type reactions. Here, only the reactant geometry is needed.
Hint
In most cases, searching transition states is not a trivial task. You may have to try several starting structures and combine NEB and dimer methods to find the transition state. The following examples are just for demonstration purposes, and may not be suitable for your own calculations.
Example: NEB and QST2/Dimer for Amine-Catalyzed Aldol Reaction
In this section, we will search the transition state of the following amine-catalyzed aldol reaction:
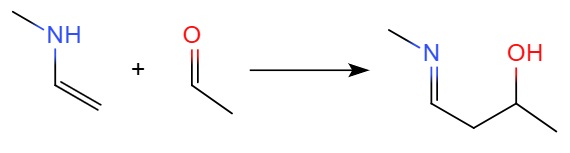
The first step is to prepare the input file for the reactant and product geometries. They are given in here:
118
2Reactant
3N -0.44812625 -0.05503381 -0.29939653
4C 0.28940454 -1.28430107 0.02594994
5C 0.42365103 0.85305301 -1.05852939
6H 1.14858130 -1.03947388 0.61482595
7H -0.34515443 -1.94528943 0.57851603
8H 0.60162891 -1.76291266 -0.87867534
9C -0.11218661 1.70819672 -1.96305927
10H 1.48128134 0.83886150 -0.89692269
11H 0.52237224 2.36918599 -2.51562444
12H -1.16981687 1.72238788 -2.12466634
13C -1.82051925 1.96886205 -0.83651451
14H -1.13776031 2.16858444 -0.03723284
15C -2.45646906 3.13519149 -1.61551609
16O -2.10383295 0.78091580 -1.13997394
17H -1.25109519 -0.28384426 -0.84974794
18H -3.37275441 3.42197565 -1.14320685
19H -1.78384110 3.96729827 -1.62387764
20H -2.65467293 2.82667229 -2.62071813
118
2Product
3N -0.22474732025333 -0.31730283410046 -0.51491194374209
4C 0.54138841877455 -1.42626714378869 0.01659619702235
5C 0.37581142979957 0.57244190213102 -1.18506998859001
6H 1.59956963538446 -1.37223656022845 -0.26069633867705
7H 0.44285772700755 -1.41562576183705 1.10277773062867
8H 0.10612456734704 -2.35145036624173 -0.36248430677077
9C -0.37357663580959 1.73548805223194 -1.76480799540763
10H 1.45524939783346 0.53941227186085 -1.38723095744134
11H 0.27481589890769 2.61572199377069 -1.78645396480960
12H -0.63716949392230 1.49356209011140 -2.80064855509307
13C -1.66735730523003 2.04995476069694 -0.98212699921514
14H -1.39146640788106 2.28056626272631 0.06585987026323
15C -2.39173812629474 3.24996769168947 -1.58779580073698
16O -2.54492763771693 0.95029342074102 -1.01960271506481
17H -2.03040745192793 0.17070983855352 -0.71586070424615
18H -3.30862690757118 3.43506393231313 -1.02954259805264
19H -1.76246901303224 4.13842282177014 -1.54692923821636
20H -2.65264077541507 3.04160762759991 -2.62544169185061
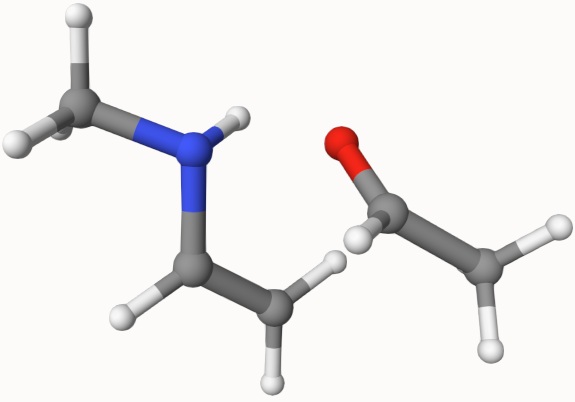
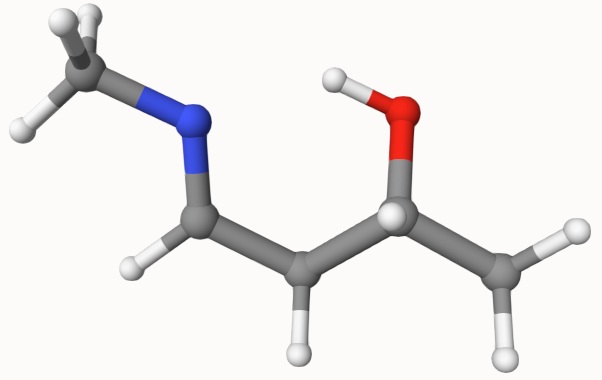
Both structures can be constructed by chemical ituition and have been optimized. However, even with these two semmingly reasonable structures, it is not easy to search the transition state. So, we will combine NEB and dimer, semiempirical and DFT methods to search the transition state.
Step 1: NEB Search. NEB is a method to search the path by connecting the reactant and product geometries. It can be a useful starting point for the dimer method. To rapidly get this, we will use xTB method:
1mol
2 r1.xyz
3end
4
5mol2
6 r2.xyz
7end
8
9basis
10 def2-svp
11end
12
13opt
14 type neb
15 num_images 15
16 neb_k 0.01
17end
18
19task
20 opt xtb
21end
Let’s examine the input file:
The reactant and product geometries are defined by
molandmol2.The NEB method is activated by
opt...endwithtype neb. The default option oftypeisminwhich is for geometry optimization, so we have to settype nebexplicitly.num_imagesis the number of images in the NEB path, which can be adjusted according to your needs. A good choice is between10and20.neb_kis the spring constant for the NEB path, which can also be adjusted according to your needs. A good choice is between0.01.
After running the input file, we can get the following output file:
anion-neb-opt.xyzThe “transition state” structure. But in many cases, we suggest you to visualize it and check whether it is reasonable.anion-neb-opt-traj.xyzThe NEB trajectory, which can be visualized by Qbics-MolStar:
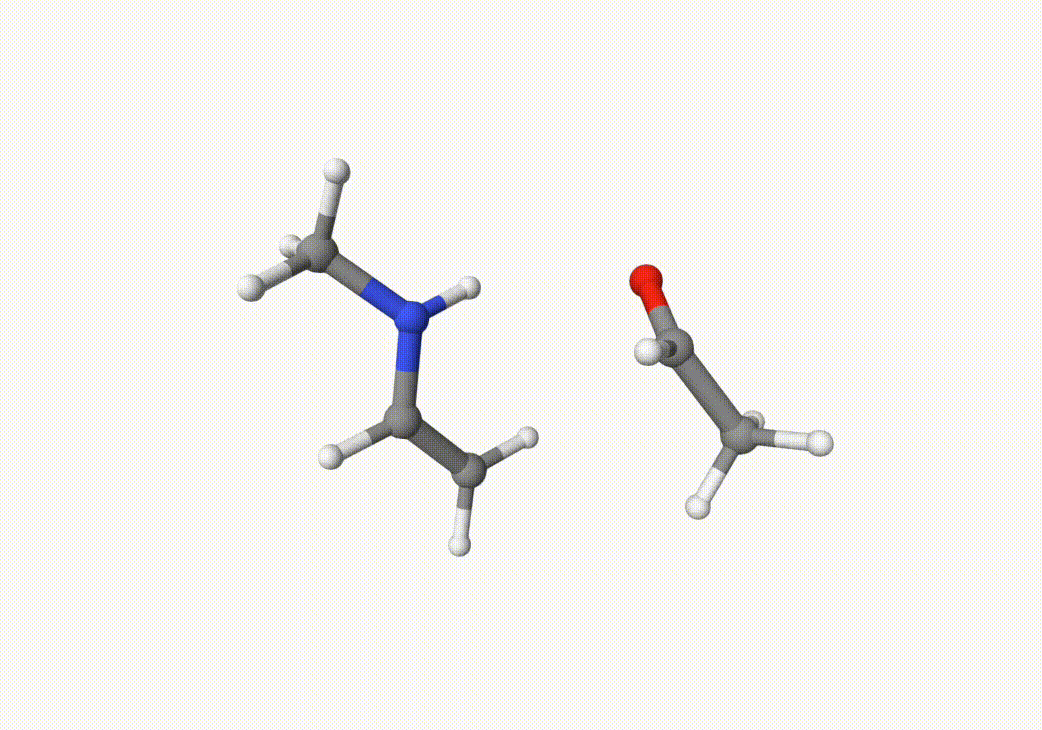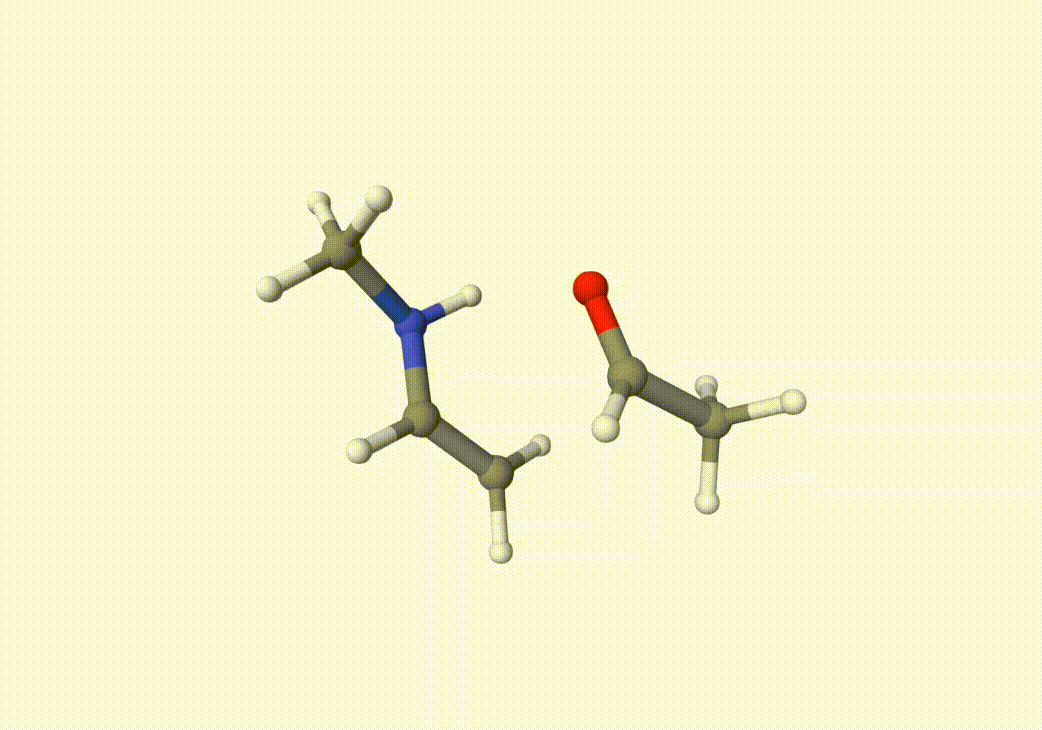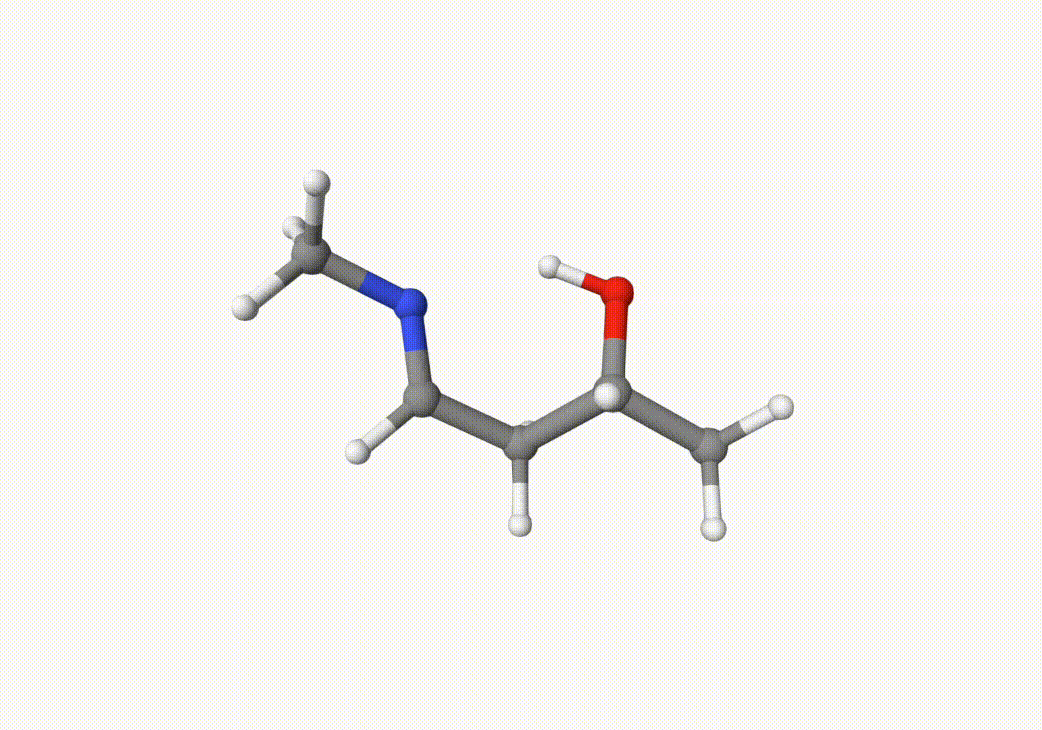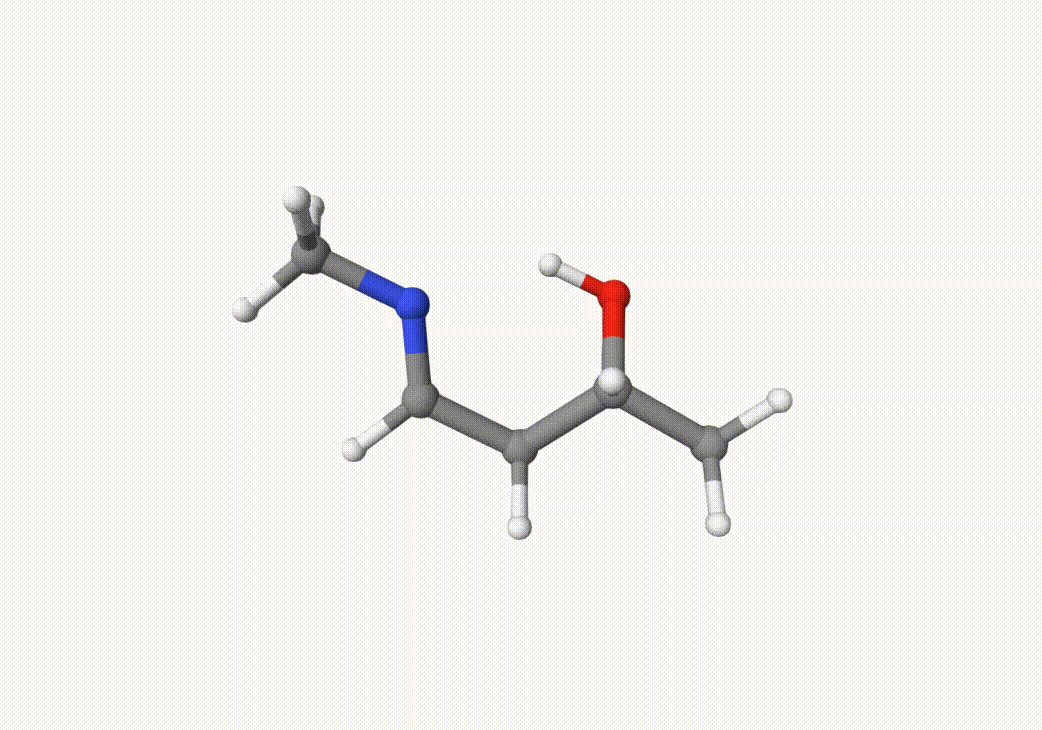
Indeed, this trajectory is our target reaction. We can choose the two closest images on the path that the trasition state may lie between them. In this case, we can choose the 5th and 7th images in anion-neb-opt-traj.xyz, which are saved in a5.xyz and a7.xyz:
118
2element x y z
3N -0.03436 -0.42780 -1.21081
4C 0.19877 -1.31325 -0.09641
5C 0.54963 0.79676 -1.27792
6H 1.21009 -1.16425 0.27978
7H -0.51219 -1.13914 0.71792
8H 0.09714 -2.34661 -0.43350
9C 0.12282 1.79024 -2.06831
10H 1.40495 0.93556 -0.62766
11H 0.65351 2.72274 -2.12058
12H -0.66378 1.63364 -2.78419
13C -1.86900 2.10882 -0.51273
14H -1.16547 2.64884 0.14563
15C -2.61217 2.99713 -1.46905
16O -2.13799 0.94532 -0.31815
17H -0.98415 -0.45631 -1.55950
18H -3.41739 3.47081 -0.90569
19H -1.97325 3.77562 -1.87311
20H -3.05645 2.41221 -2.27009
118
2element x y z
3N -0.04991 -0.40765 -1.08610
4C 0.35194 -1.39200 -0.13587
5C 0.50298 0.72490 -1.16520
6H 1.36153 -1.23389 0.25614
7H -0.36195 -1.32388 0.69157
8H 0.25447 -2.38320 -0.57375
9C -0.26333 1.81516 -1.81912
10H 1.42373 0.97712 -0.62468
11H 0.32790 2.72274 -1.93602
12H -0.65750 1.50523 -2.78663
13C -1.50488 2.07052 -0.89479
14H -1.14590 2.52877 0.04533
15C -2.47962 3.02762 -1.58061
16O -2.17108 0.87844 -0.60177
17H -1.60693 0.12971 -0.93634
18H -3.25074 3.33649 -0.87990
19H -1.95994 3.90581 -1.95044
20H -2.96008 2.50844 -2.40618
Now we will use the dimer/QST2 algorithm to search the transition state between these two images. Below, we show how to do this on B3LYP/def2-SVP levels of theory:
1mol
2 a5.xyz
3end
4
5mol2
6 a7.xyz
7end
8
9basis
10 def2-svp
11end
12
13scf
14 charge 0
15 spin2p1 1
16end
17
18opt
19 type qst2 # You can also use dimer.
20end
21
22task
23 opt b3lyp
24end
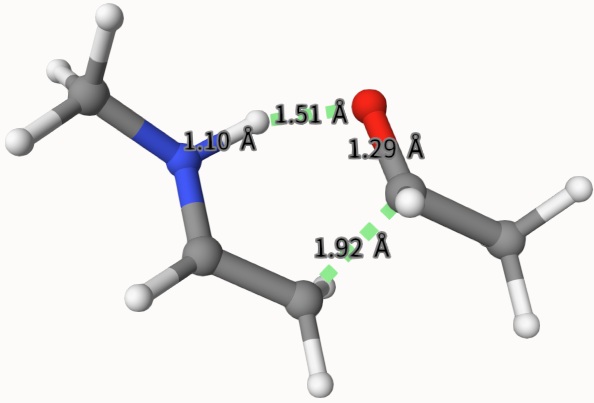
Fortunately, we can get the transition state structure, which is saved in anion-dft-qst2-opt.xyz and is shown below:
Example: dMECP for F-+CH3Cl = CH3F+Cl-
For AB+C=A+BC type reactions, we can use the dMECP method to search the transition state. For example, for the reaction F-+CH3Cl = CH3F+Cl-, we can use the following input file:
1basis
2 def2-svp
3end
4
5mol
6 C -0.43654823 1.13197968 0.00000000
7 H -0.07987539 1.63637787 0.87365150
8 H -0.07987539 1.63637787 -0.87365150
9 H -1.50654823 1.13199286 0.00000000
10 Cl 0.15009830 -0.52737135 0.00000000
11 F -1.63124586 2.75454539 -0.44024554
12end
13
14scf
15 type u # This must be set.
16 charge -1
17 spin2p1 1
18end
19
20mecp
21 num_steps 200
22 energy_cov 1.E-5
23 # Reactant.
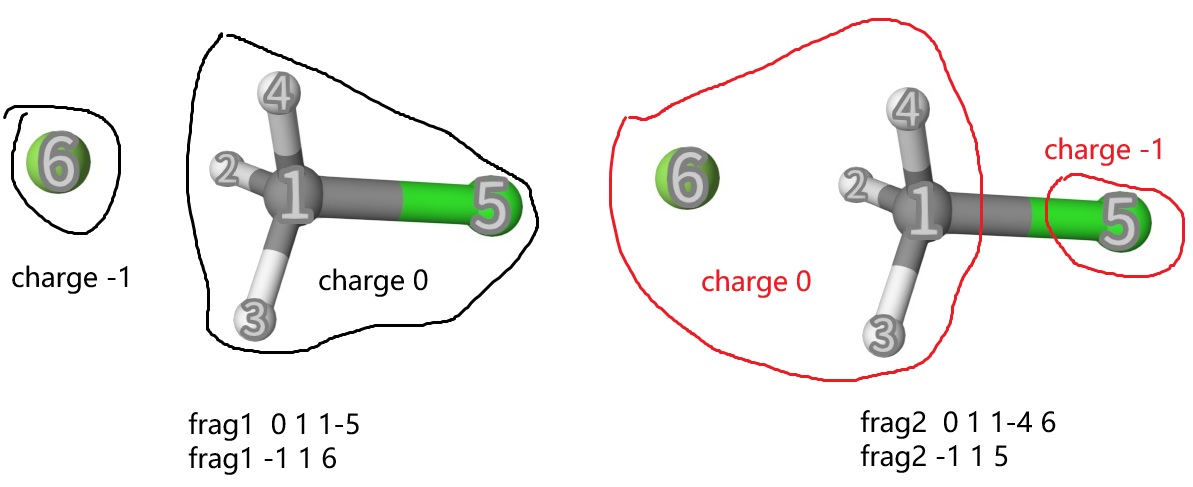
24 frag1 0 1 1-5
25 frag1 -1 1 6
26 # Product.
27 frag2 0 1 1-4 6
28 frag2 -1 1 5
29end
30
31task
32 dmecp b3lyp
33end
We can see that we want to search the transition state at B3LYP/def2-SVP level of theory. The dMECP method is activated by dmecp in task...end, and is controlled by mecp...end option. Below are some important points:
dMECP is implemented with the TSO method, so in
scf...end, we have to settype u.num_stepsis the number of steps for the dMECP search, which can be adjusted according to your needs.The reactant and product connectivity are defined by
frag1andfrag2, respectively. Their grammar is similar tofragin eda and scfguess, but here we have to define the connectivity of the reactant and product separately byfrag1andfrag2. The geometry structure infch3cl.inpis shown below:
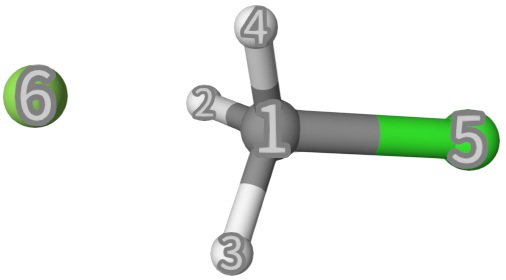
The frag1 and frag2 definitions are shown below:
Therefore, we just need to define the “connectivity” and “charge” of the reactant and product, no matter what the geometry is. The dMECP method will automatically optimize the geometry to find the transition state. dMECP only needs reactant complex struture. This is very convenient for the transition state search.
After running the input file, we can get the following output file:
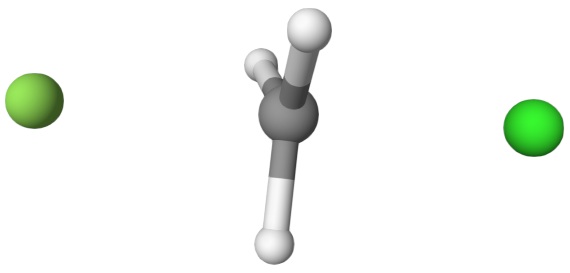
fch3cl-mecp.xyzThe final transition state.fch3cl-mecp-traj.xyz. The dMECP trajectory.
Both files can be visualized by Qbics-MolStar:
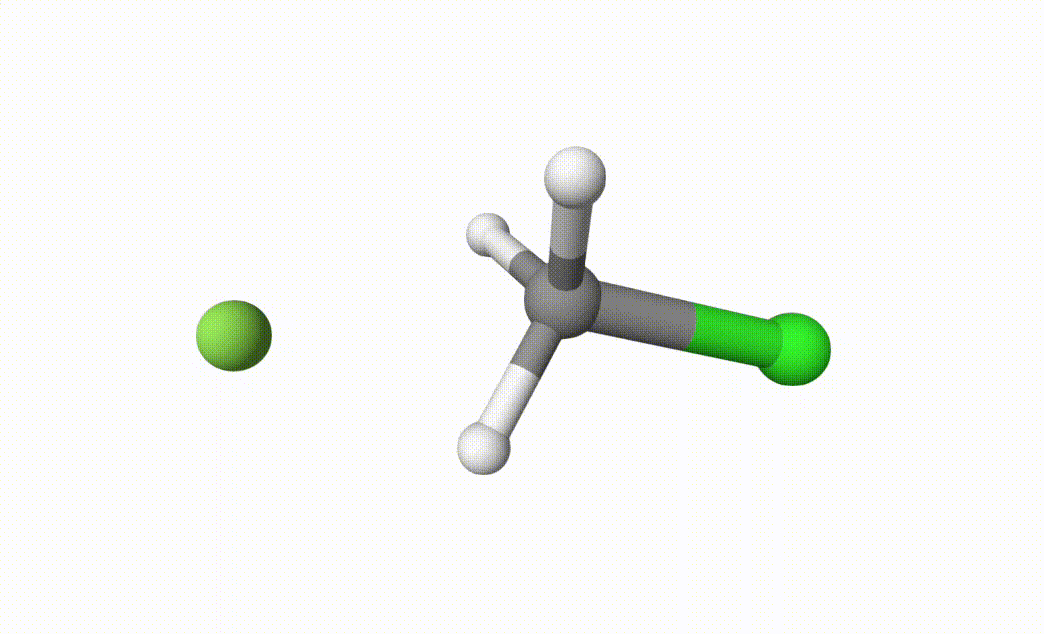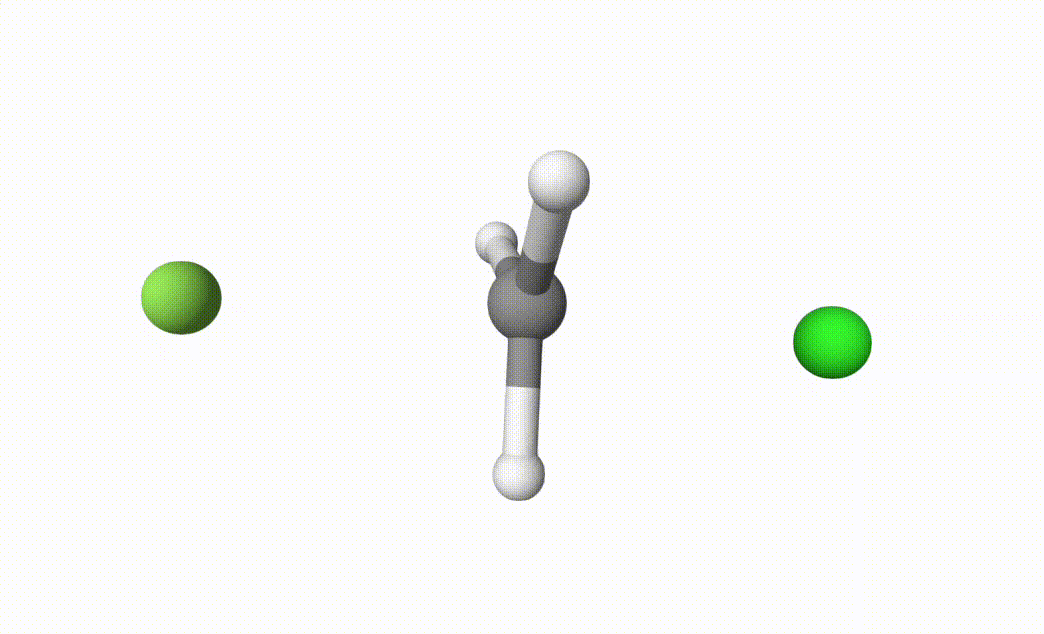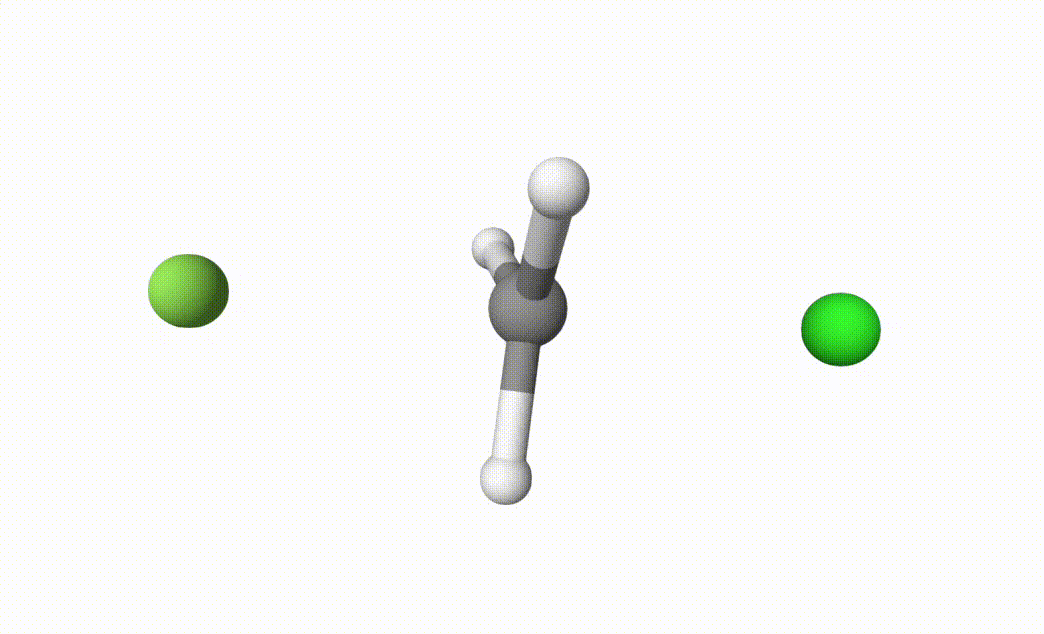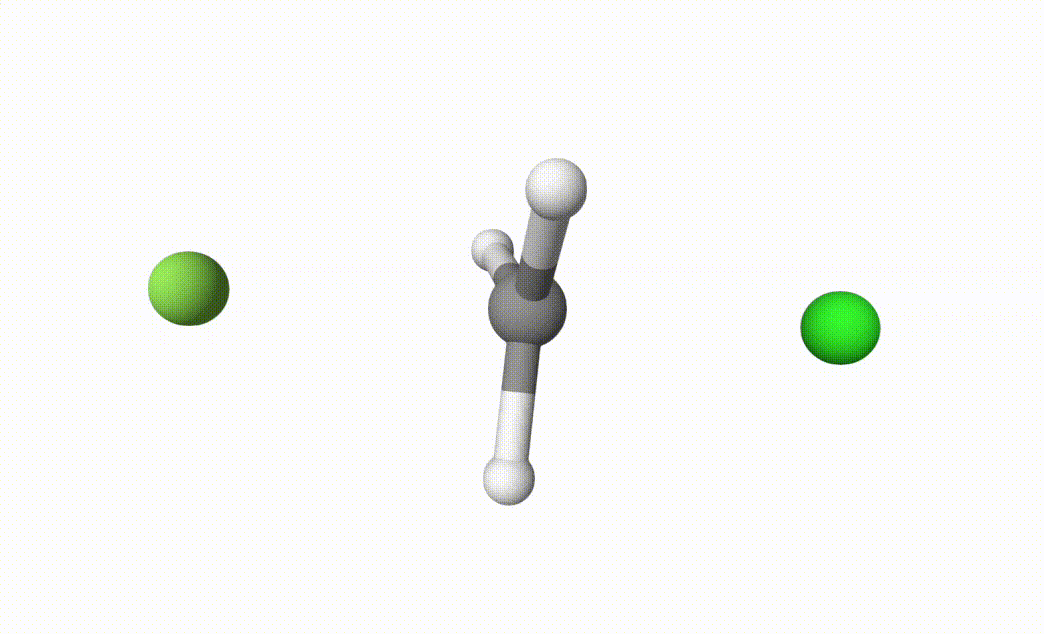
In fch3cl.out, we can find following content:
1Adiabatic state energy: -599.69936976 Hartree
2Diabatic state 1 energy: -599.62911003 Hartree
3Diabatic state 2 energy: -599.62485338 Hartree
4|E(2)-E(1)|: 0.00425665 Hartree
5delta|E(2)-E(1)|: 0.00000520 Hartree
6RMS gradient: 0.00008376 Hartree/Bohr
7RMS displacement: 0.00098426 Angstrom
Here, Adiabatic state energy is the energy of standard DFT. Diabatic state 1 energy and Diabatic state 2 energy is the diabatic energy of frag1 and frag2.