Tip
All input files can be downloaded: Files.
Tip
For more information of this section, please refer to these keywords:
TSO-DFT (1): Excited States
This tutorial will lead you step by step to do target state optimization DFT (TSO-DFT) using Qbics. TSO-DFT is an originally developed method in Qbics. It is a powerful and accurate method for excited and diabatic states. In many physical processes like charge transfer, TSO-DFT can give much more reasonable results than TDDFT. Therefore, it is worth learning this method.
Hint
If you use Qbics to do TSO-DFT in you paper, please cite the following references:
In this tutorial, we will introduce how to use TSO-DFT to study exicted states.
Example: HCHO n→π* Excitation Energy
In the first example of TSO-DFT, we will show how to calculate the n→π* excitation energy of HCHO at B3LYP/cc-pVTZ level of theory.
Hint
Accurate calculations of excited states need basis sets of high quality. We recommend cc-pVTZ or beyond for energy evaluation and cc-pVDZ or beyond for geometry optimization.
Ground State
Before studying excited states, it is always beneficial to look at the ground state first. The input for ground state is:
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type R
9end
10
11mol
12 O -0.68710791 0.04339108 0.00000000
13 C 0.50595906 -0.07524732 0.00000008
14 H 1.09294418 -0.13258299 -0.93605972
15 H 1.09294415 -0.13258317 0.93605964
16end
17
18task
19 energy b3lyp
20end
This is a standard SCF calculation. After calculation, we will obtain the output file hcho-gs.out and wavefunction file hcho-gs.mwfn. In hcho-gs.out, we can find these:
1SCF Structure:
2 # of electrons: 16
3 # of alpha electrons: 8
4 # of beta electrons: 8
5 2S+1: 1
6 Spin alignment: Restricted
7 Temperature: 0.00000000 Kelvin
8 # of basis functions: 88
Thus, this system contains 16 electrons and 88 orbitals. We will calculate the excited states based on this information.
Excited State
The n→π* excited state is a state that an electron is excited from orbital 8 to 9. To calculate this state, one can use one of these 2 input files:
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type U # This is needed for TSO-DFT.
9 no_scf tso
10end
11
12scfguess
13 type mwfn
14 file hcho-gs.mwfn
15 orb 16 1 1-87 : 1-7 9-88
16 orb 0 1 88 : 8
17end
18
19mol
20 O -0.68710791 0.04339108 0.00000000
21 C 0.50595906 -0.07524732 0.00000008
22 H 1.09294418 -0.13258299 -0.93605972
23 H 1.09294415 -0.13258317 0.93605964
24end
25
26task
27 energy b3lyp
28end
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type U # This is needed for TSO-DFT.
9 no_scf tso
10end
11
12scfguess
13 type tso
14 frag 0 1 1-4
15 orb 16 1 1-87 : 1-7 9-88
16 orb 0 1 88 : 8
17end
18
19mol
20 O -0.68710791 0.04339108 0.00000000
21 C 0.50595906 -0.07524732 0.00000008
22 H 1.09294418 -0.13258299 -0.93605972
23 H 1.09294415 -0.13258317 0.93605964
24end
25
26task
27 energy b3lyp
28end
First, we claim that hcho-ee-1.inp and hcho-ee-2.inp have the same task: calculating n→π* excited state. They differ at type in scfguess...end:
In
hcho-ee-1.inp,type mwfnmeans that the reference state is given by a MWFN file, whose name isfile hcho-gs.mwfn, that is, the ground state calculated above.In
hcho-ee-2.inp,type tsomeans that the reference state is calculated automatically by Qbics. The calculated reference state will be saved tohcho-ee-2-ref.mwfn. The reference state is given by the followingfrag 0 1 1-4. Thisfragis to assign the whole molecule (atom1-4) with charge0and spin multiplicity1as reference. See scfguess for more information.
In both hcho-ee-1.inp and hcho-ee-2.inp, the excited state is assigned by orb. In TSO-DFT, the most important concept is the partition of orbital subspace, see below:
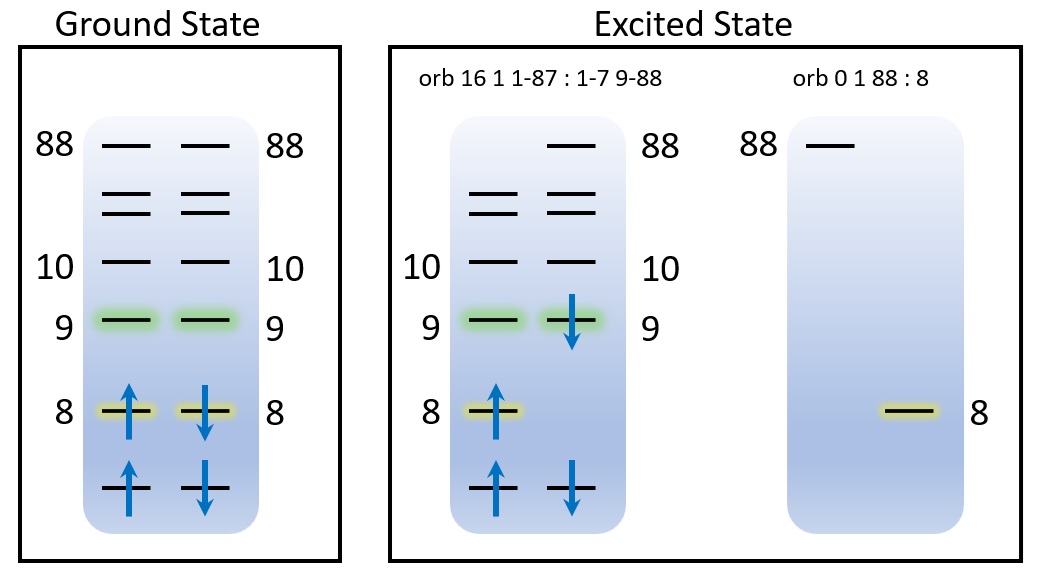
As shown by this picture, in the ground state, the alpha and beta orbitals are occupied by 8 electrons, respectively. In the n→π* excited state, i.e., the state that an electron is excited from orbital 8 to 9, we partition orbitals into 2 subspaces (2 orb):
orb 16 1 1-87 : 1-7 9-88The first subspace contains16electrons and spin multiplicity is1. The numbers separated by:are the alpha orbital indices1-87and beta orbital indices1-7 9-88, respecitively. Here, we remove beta orbital 8, so when the electrons are occupied, it will occupy9automatically and beta8is never occupied. So, an n→π* excited state is automatically constructed. Note that we also remove an alpha orbital87, since in each orbital subspace, the number of alpha and beta orbital must be the same. Therefore, we remove the highest one.orb 0 1 87 : 8The second subspace it the complementary set of the first one, so it is zero-occupied.
Now we can calculate the excited states. After calculation, we will obtain the excited state wavefunction hcho-ee-1.mwfn or hcho-ee-2.mwfn. The excited state properties can be found in hcho-ee-1.out or hcho-ee-2.out:
1TSO Transition
2==============
3Reference wave function read from: hcho-ee-2-ref.mwfn
4Reference energy: -114.54951400 Hartree
5Current energy: -114.42013176 Hartree
6E(Current)-E(Ref): 3.52067019 eV
7Transition dipole moment (Debye): 0.00004 -0.00000 -0.00112
8Oscillator strength: 0.00000
9Higher order corrections:
10Transition quadrupole moment (Debye*Angstrom):
11 Qxx: -0.00001; Qyy: 0.00003; Qzz: -0.00002
12 Qxy: 0.00000; Qxz: -0.06437; Qyz: -0.63367
13Quadrupole correction to oscillator strength: 4.31049E-09
14Transition angular momentum (au): 0.06258 -0.00609 -0.00032
15Magnetic dipole correction to oscillator strength: 4.53975E-09
Thus, the excited energy is 3.52 eV. The oscillator strength is 0.00, suggesting that this is a dark state.
Example: HCHO n2→(π*)2 Excitation Energy
Now we consider a doubly excited state, where two electons are excited from n to π*.
Hint
The popular TDDFT code in most programs are implemented with adiabatic approximation, making TDDFT unable to calculate double excitation. This is also a reason that you should use TSO-DFT: due to the theoretical flaws in TDDFT, many types of excitation, like double, core, and charge transfer excitations, cannot be studies by TDDFT, but all of them can be studied by TSO-DFT with balanced accuracy. For theoretical details, please refer to:
To calculate this n2→(π*)2 excitation, we can use one of these 2 files:
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type U # This is needed for TSO-DFT.
9 no_scf tso
10end
11
12scfguess
13 type mwfn
14 file hcho-gs.mwfn
15 orb 16 1 1-7 9-88 : 1-7 9-88
16 orb 0 1 8 : 8
17end
18
19mol
20 O -0.68710791 0.04339108 0.00000000
21 C 0.50595906 -0.07524732 0.00000008
22 H 1.09294418 -0.13258299 -0.93605972
23 H 1.09294415 -0.13258317 0.93605964
24end
25
26task
27 energy b3lyp
28end
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type U # This is needed for TSO-DFT.
9 no_scf tso
10end
11
12scfguess
13 type tso
14 frag 0 1 1-4
15 orb 16 1 1-7 9-88 : 1-7 9-88
16 orb 0 1 8 : 8
17end
18
19mol
20 O -0.68710791 0.04339108 0.00000000
21 C 0.50595906 -0.07524732 0.00000008
22 H 1.09294418 -0.13258299 -0.93605972
23 H 1.09294415 -0.13258317 0.93605964
24end
25
26task
27 energy b3lyp
28end
These input files are very similar to the ones for n→π* excited state:
In
hcho-de-1.inp,type mwfnmeans that the reference state is given by a MWFN file, whose name isfile hcho-gs.mwfn, that is, the ground state calculated above.In
hcho-de-2.inp,type tsomeans that the reference state is calculated automatically by Qbics. The calculated reference state will be saved tohcho-de-2-ref.mwfn. The reference state is given by the followingfrag 0 1 1-4. Thisfragis to assign the whole molecule (atom1-4) with charge0and spin multiplicity1as reference. See scfguess for more information.
The orbital partition is given below:
orb 16 1 1-7 9-88 : 1-7 9-88The first subspace contains16electrons and spin multiplicity is1. The numbers separated by:are the alpha orbital indices1-7 9-88and beta orbital indices1-7 9-88, respecitively. Here, we remove alpha/beta orbital 8, so when the electrons are occupied, they will occupy alpha/beta9automatically and alpha/beta8is never occupied. So, an n2→(π*)2 doubly excited state is automatically constructed.orb 0 1 8 : 8The second subspace it the complementary set of the first one, so it is zero-occupied.
Now we can calculate the excited states. After calculation, we will obtain the excited state wavefunction hcho-de-1.mwfn or hcho-de-2.mwfn. The excited state properties can be found in hcho-de-1.out or hcho-de-2.out:
1TSO Transition
2==============
3Reference wave function read from: hcho-de-2-ref.mwfn
4Reference energy: -114.54951400 Hartree
5Current energy: -114.15687470 Hartree
6E(Current)-E(Ref): 10.68425953 eV
7Transition dipole moment (Debye): 0.00000 0.00000 0.00000
8Oscillator strength: 0.00000
9Higher order corrections:
10Transition quadrupole moment (Debye*Angstrom):
11 Qxx: 0.00000; Qyy: 0.00000; Qzz: 0.00000
12 Qxy: 0.00000; Qxz: 0.00000; Qyz: 0.00000
13Quadrupole correction to oscillator strength: 0.00000E+00
14Transition angular momentum (au): 0.00000 0.00000 0.00000
15Magnetic dipole correction to oscillator strength: 0.00000E+00
Thus, the excited energy is 10.68 eV.
Example: HCHO n→π* Excited State Geometry
Geometry optimization of excited states by TSO-DFT is very easy. Just remember the following 3 points:
Geometry optimization by TSO-DFT can only use
type tsoinscfguess...end;Replace
energytoopt;Sometimes, excited state geometry have completely different symmtery from the ground state, in this case, some manually symmetry broken is needed.
The first 2 points are easy to understand. The third point will be explained by examples.
Now we want to optimize the structure of n→π* excited state of HCHO. We just need to replace energy to opt in hcho-ee-2.inp. We will call this file hcho-ee-geom-1.inp:
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type U # This is needed for TSO-DFT.
9 no_scf tso
10end
11
12scfguess
13 type tso
14 frag 0 1 1-4
15 orb 16 1 1-87 : 1-7 9-88
16 orb 0 1 88 : 8
17end
18
19mol
20 O -0.68710791 0.04339108 0.00000000
21 C 0.50595906 -0.07524732 0.00000008
22 H 1.09294418 -0.13258299 -0.93605972
23 H 1.09294415 -0.13258317 0.93605964
24task
25 opt b3lyp
26end
This file will of course do the geometry optimization for excited state of HCHO. However, note that the initial geometry is a planar one, so geometry optimization will always keep this planarity. The n→π* excitation will lead to a broken of planarity of molecules, so we should give this molecule a perturbation to enable geometry optimization to treat non-planar molecules. This is given in hcho-ee-geom-2.inp:
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type U # This is needed for TSO-DFT.
9 no_scf tso
10end
11
12scfguess
13 type tso
14 frag 0 1 1-4
15 orb 16 1 1-87 : 1-7 9-88
16 orb 0 1 88 : 8
17end
18
19mol
20 O -0.68710791 0.24339108 0.00000000 # Move O atom
21 C 0.50595906 -0.27524732 0.00000008 # Move C atom
22 H 1.09294418 -0.13258299 -0.93605972
23 H 1.09294415 -0.13258317 0.93605964
24end
25
26task
27 opt b3lyp
28end
You can see that, we let C and O move out of the molecular plane by about 0.2 Angstrom.
After calculations, we can check the optimized geometry of hcho-ee-geom-1-opt.xyz and hcho-ee-geom-2-opt.xyz with Qbics-MolStar:
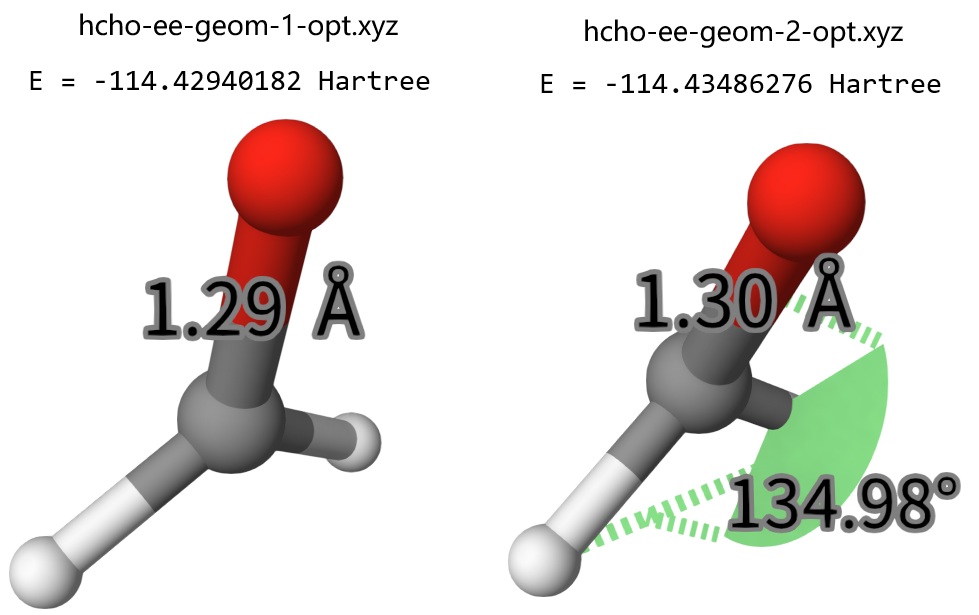
We found that, the second non-planar geometry is more stable than the planar one by 0.0054 Hartree. Interestingly, if we optimize the non-planar HCHO at ground state, it will return to the planar one. Anyway, since the potential energy surface of excited states can have completely different topology compared to the ground state one, you must take care if your excited state structure is a global minimum. Below is another example.
Example: HCCH Non-linear Excited State Geometry
HCCH is a linear molecule. Its ground state geometry can be easily determined. Now, we will determine its geometry for the HOMO→LUMO excited state, which is called S1 state.
First, the initial structure is the ground state minimum:
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type U # This is needed for TSO-DFT.
9 no_scf tso
10end
11
12scfguess
13 type tso
14 frag 0 1 1-4
15 orb 14 1 1-87 : 1-6 8-88
16 orb 0 1 88 : 7
17end
18
19mol
20 H 0.0 0.0 -1.65969619
21 C 0.0 0.0 -0.59806842
22 C 0.0 0.0 0.59805611
23 H 0.0 0.0 1.65970850
24end
25
26task
27 opt b3lyp
28end
However, with a linear initial guess, the local geometry optimization process can only lead to a linear molecule. Chemical intuition tells us that in the excited states, HCCH may bend. So, let’s consider 2 cases: the 2 hydrogen atoms are in cis and trans geometry. In the input file, we will simply move the 2 hydrogen atoms in the same and opposite direction, respecitvely, by a small distance:
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type U # This is needed for TSO-DFT.
9 no_scf tso
10end
11
12scfguess
13 type tso
14 frag 0 1 1-4
15 orb 14 1 1-87 : 1-6 8-88
16 orb 0 1 88 : 7
17end
18
19mol
20 H 0.5 0.5 -1.65969619 # Move H in the same direction
21 C 0.0 0.0 -0.59806842
22 C 0.0 0.0 0.59805611
23 H 0.5 0.5 1.65970850 # Move H in the same direction
24end
25
26task
27 opt b3lyp
28end
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type U # This is needed for TSO-DFT.
9 no_scf tso
10end
11
12scfguess
13 type tso
14 frag 0 1 1-4
15 orb 14 1 1-87 : 1-6 8-88
16 orb 0 1 88 : 7
17end
18
19mol
20 H 0.5 0.5 -1.65969619 # Move H in opposite direction
21 C 0.0 0.0 -0.59806842
22 C 0.0 0.0 0.59805611
23 H 0.5 -0.5 1.65970850 # Move H in opposite direction
24end
25
26task
27 opt b3lyp
28end
After calculations, we collect the optimized structures in hcch-1-opt.xyz, hcch-2-opt.xyz, and hcch-3-opt.xyz, shown below:
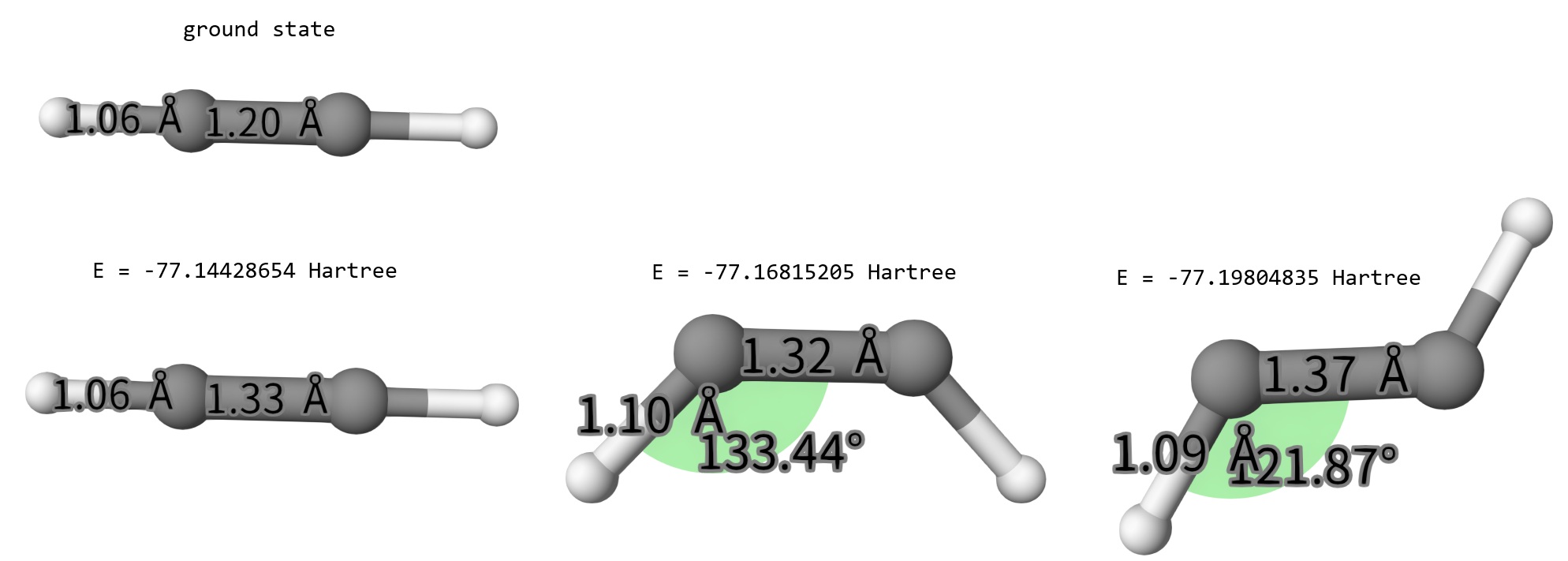
Obviously, the linear geometry is far from global optimization, being completely different from the ground state. In S1 state, the trans isomer is the most stable one.
This example again emphasizes the compexity of excited state potential energy surfac! If you start the geometry optimization with a structure of high symmetry, you may NEVER arrive at the true minimum, so try more initial structure!