Tip
All input files can be downloaded: Files.
opt
This keyword controls the search of geometry minimum, transistion state, and reaction path.
Options
- type
Value
MinSearch the geometry minimum.NEBUse the nudged elastic band (NEB) algorithm to search the reaction path and transition state.DimerUse the dimer algorithm to search the transition state given the reactant and product geometry.QST2Use the QST2 algorithm to search the transition state given the reactant and product geometry.TSSearch the transition state from a single structure.Default
MinDetermine what type of geometry optimization you want to do.
For
MinandTS, the initial structure should be given inmol. The optimization process is output tox-opt-traj.xyzand the final minimum or transition state is output tox-opt.xyz.For
NEB,Dimer, andQST2, 2 structures have to be given inmolandmol2, which represent a reactant and product pose, respectively. Usually, one can first useNEBto rapidly find a reasonable path and transition state. IfNEBis hard to converge, then use the structures fromNEBresult to do aDimerorQST2search.DimerandQST2require reactant and product structures of high quality.For
NEB, the reaction path is output tox-opt-traj.xyzand the final transition state is output tox-opt.xyz.For
DimerandQST2, the transition state is output tox-opt.xyz. Thex-opt-traj.xyzis NOT reaction path! It is just the optimization process asMinorTS.
- max_step
Value
An integer
Default
1000The maximum number of geometry optimization steps.
- energy_cov
Value
A real number
Default
1.E-4The energy convergence threshold. When the energy change is smaller than this value, this energy condition is satisfied.
- grad_cov
Value
A real number
Default
1.E-3The gradient convergence threshold. This actually determines 4 convergence thresholds:
Maximum gradient component
grad_covRMS gradient:
grad_cov* 2/3Maximum atomic displacement
grad_cov* 4RMS atomic displacement
grad_cov* 8/3When all these 4 conditions are met, this gradient condition is satisfied.
- max_dr
Value
A real number
Default
0.5The maximum atomic displacement in an optimization step. If the molecule is highly flexible (Mathematically, the potential energy surface is very flat), or the structure (especially transition state) is very close to the stationary point but not converge, setting a smaller
max_drlike0.1is very useful.
- num_images
Value
An integer
Default
10The number of images for NEB transition state search. This number canNOT be set too small, say,
5.
- neb_k
Value
A real number
Default
0.01The force constant for NEB transition state search. For a specific system, the optimal number of
neb_kshould be chose by trail-and-error.
- fix_atoms
Value
Atom range
Default
None
Assign the atoms that are fixed during geometry optimization. For example:
1opt 2 fix_atom 2 5-9 23 26 3end
The atoms 2,5,6,7,8,9,23,26 will be fixed during geometry optimization.
- fix_bond
Value
2 integers
Default
None
Assign the bond that are fixed during geometry optimization. For example:
1opt 2 fix_bond 1 4 3 fix_bond 2 6 4end
The bonds (1,4) and (2,6) will be fixed during geometry optimization.
- fix_angle
Value
3 integers
Default
None
Assign the angle that are fixed during geometry optimization. For example:
1opt 2 fix_angle 1 4 5 3 fix_angle 2 6 7 4end
The angles (1, 4, 5) and (2, 6, 7) will be fixed during geometry optimization.
- fix_torsion
Value
4 integers
Default
None
Assign the torsion that are fixed during geometry optimization. For example:
1opt 2 fix_torsion 1 4 5 9 3 fix_torsion 2 6 7 12 4end
The torsions (1, 4, 5, 9) and (2, 6, 7, 12) will be fixed during geometry optimization.
Attention
Currently, the transition state search algorithm QST2 and TS do NOT support constraints. If you want to search a transition state with constraints, please use NEB or Dimer.
Theoretical Background
Minimum
The minimum is defined as a stable isomer on its potential energy surface (PES) of a molecule. The gradients on all atoms are zero. The optimization of minimum depends strongly on the initial structure. For different starters, one can get different isomers.
Transition State
Transition state is a short-lived configuration of atoms that in maximum on one direction but minimum on other directions. The gradients on all atoms are also zero. In Qbics, one can use NEB or dimer method to search the transition state. Only (unoptimized) reactant and product structures are needed. No exact Hessian needs to be computed.
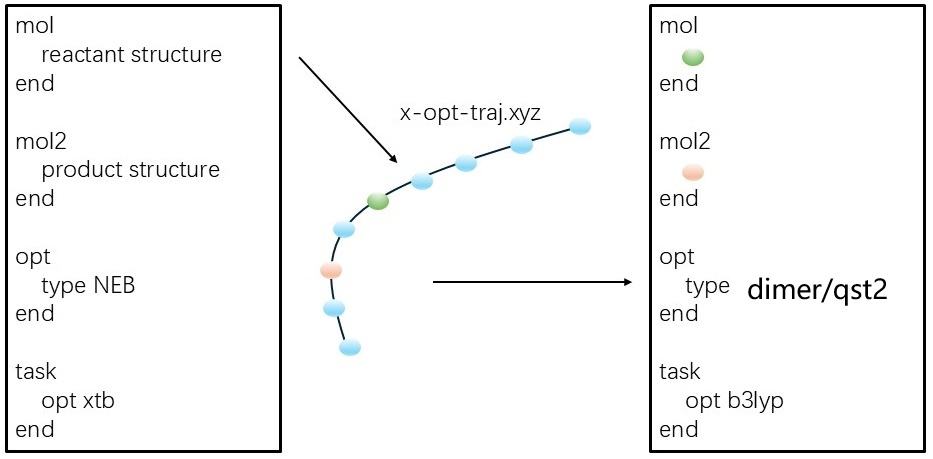
A good strategy is:
Use cheap method, like xTB, to find a reasonable path and transition state with NEB (
type neb).Then, use standard DFT method to refine the transition state with dimer (
type dimer) or QST2 (type QST2), even the previous NEB result is not converged.
This strategy is shown below:
Input Examples
Example: Minimum Structure of Aspirin
Search the minimum structure of aspirin at B3LYP/def2-SVP level of theory:
1basis
2 def2-svp
3end
4
5scf
6 charge 0
7 spin2p1 1
8end
9
10mol
11 O 1.23330 0.55400 0.77920
12 O -0.69520 -2.71480 -0.75020
13 O 0.79580 -2.18430 0.86850
14 O 1.78130 0.81050 -1.48210
15 C -0.08570 0.60880 0.44030
16 C -0.79270 -0.55150 0.12440
17 C -0.72880 1.84640 0.41330
18 C -2.14260 -0.47410 -0.21840
19 C -2.07870 1.92380 0.07060
20 C -2.78550 0.76360 -0.24530
21 C -0.14090 -1.85360 0.14770
22 C 2.10940 0.67150 -0.31130
23 C 3.53050 0.59960 0.16350
24 H -0.18510 2.75450 0.65930
25 H -2.72470 -1.36050 -0.45640
26 H -2.57970 2.88720 0.05060
27 H -3.83740 0.82380 -0.50900
28 H 3.72900 1.41840 0.85930
29 H 4.20450 0.69690 -0.69240
30 H 3.71050 -0.36590 0.64260
31 H -0.25550 -3.59160 -0.73370
32end
33
34task
35 opt b3lyp
36end
Example: Minimum Structure with Constraints
Search the minimum structure of a molecule “1UML” with a bond and a torsion fixed at xTB level of theory:
1opt
2 fix_bond 7 51
3 fix_torsion 2 9 12 13
4end
5
6mol
7 1uml.xyz
8end
9
10task
11 opt xtb
12end
1 60
21UMl(0)
3 C 0.38066735 2.55219474 1.46267636
4 N -0.33789766 3.82795956 1.53541195
5 C -1.09121603 3.87593593 2.64608144
6 N -0.94245652 2.73117700 3.32171417
7 C -0.03158842 1.89319979 2.55453525
8 C 0.37212443 0.52262484 2.93349994
9 O 0.95733416 -0.14814113 2.10415165
10 N 0.07166899 0.06707847 4.15265909
11 C -0.24927084 4.91745505 0.56953160
12 C 0.93256260 5.84926075 0.89138538
13 O 0.85638839 6.40073825 2.18023567
14 C -0.21178237 4.41479892 -0.88585063
15 C -1.30451123 3.40488529 -1.25181254
16 N -0.92687172 2.67905416 -2.46663252
17 C -1.02012131 3.09677073 -3.75965819
18 C -0.51157942 2.12142921 -4.62384570
19 C -0.09088312 1.03387236 -3.77904056
20 C -0.36650560 1.41729423 -2.43324566
21 C 0.47064744 -0.18584082 -4.05073192
22 C 0.77451739 -1.04742178 -2.99644766
23 C 0.51501197 -0.67068355 -1.66606604
24 C -0.06269010 0.57406126 -1.40267529
25 N 0.81935074 -1.45803325 -0.60027087
26 C 1.28805430 -2.72903213 -0.64795569
27 O 1.47475540 -3.39721237 -1.66461437
28 C 1.57993549 -3.25675705 0.74748896
29 C 1.64225207 -4.78720972 0.77417719
30 C -0.79428279 -4.92640828 1.45233103
31 C 0.27286077 -5.43783743 0.71500759
32 C 0.09621230 -6.58712911 -0.05216185
33 C -1.13149600 -7.23287171 -0.06650889
34 C -2.18992415 -6.73793686 0.69577410
35 C -2.02072844 -5.58468177 1.44785223
36 H 1.08446564 2.21827120 0.70020968
37 H -1.72176781 4.71224102 2.94804163
38 H -0.42535733 0.67320222 4.82086735
39 H 0.33691344 -0.89030993 4.42449590
40 H -1.16218006 5.52526169 0.70631806
41 H 0.98220194 6.64417179 0.12373872
42 H 1.88374201 5.29787879 0.83772723
43 H 0.29674389 7.26495989 2.15187259
44 H 0.77353707 3.94775022 -1.06475242
45 H -0.26304168 5.28737023 -1.55851059
46 H -2.24791634 3.94699892 -1.42947786
47 H -1.48837270 2.67328764 -0.45339564
48 H -1.43235349 4.05491839 -4.07609186
49 H -0.44710423 2.17375337 -5.71065901
50 H 0.67778198 -0.47993189 -5.07960408
51 H 1.21706689 -2.02142178 -3.20534792
52 H -0.27079576 0.87203324 -0.37507411
53 H 0.68015960 -1.04879454 0.33459425
54 H 0.94277827 -2.86688017 1.55772521
55 H 2.59161906 -2.87935288 0.99367456
56 H 2.12787894 -5.07775312 1.72353104
57 H 2.29189074 -5.17230270 -0.02720370
58 H -0.66818790 -4.01186770 2.03180634
59 H 0.92422969 -6.97980706 -0.64230710
60 H -1.26922150 -8.12751976 -0.67371391
61 H -3.14853209 -7.25672910 0.70037925
62 H -2.85015656 -5.19299484 2.03675744
Check the constraints during optimization:
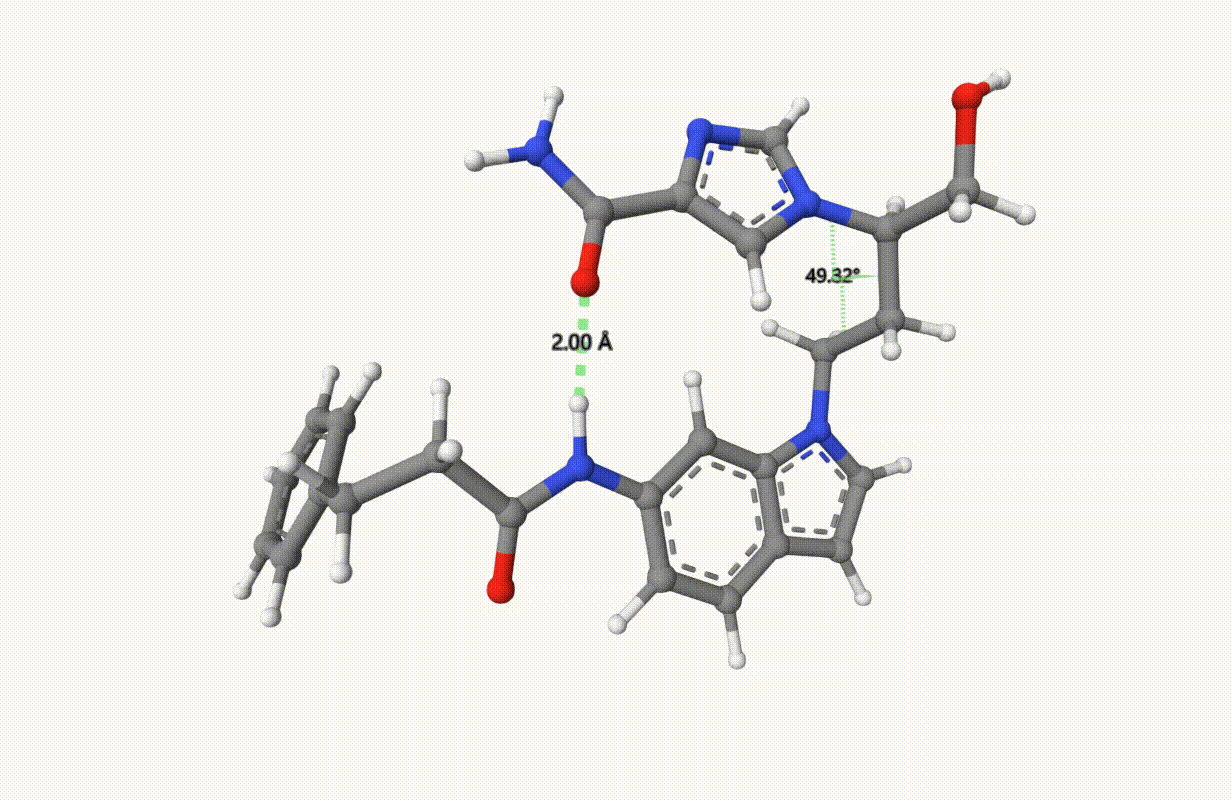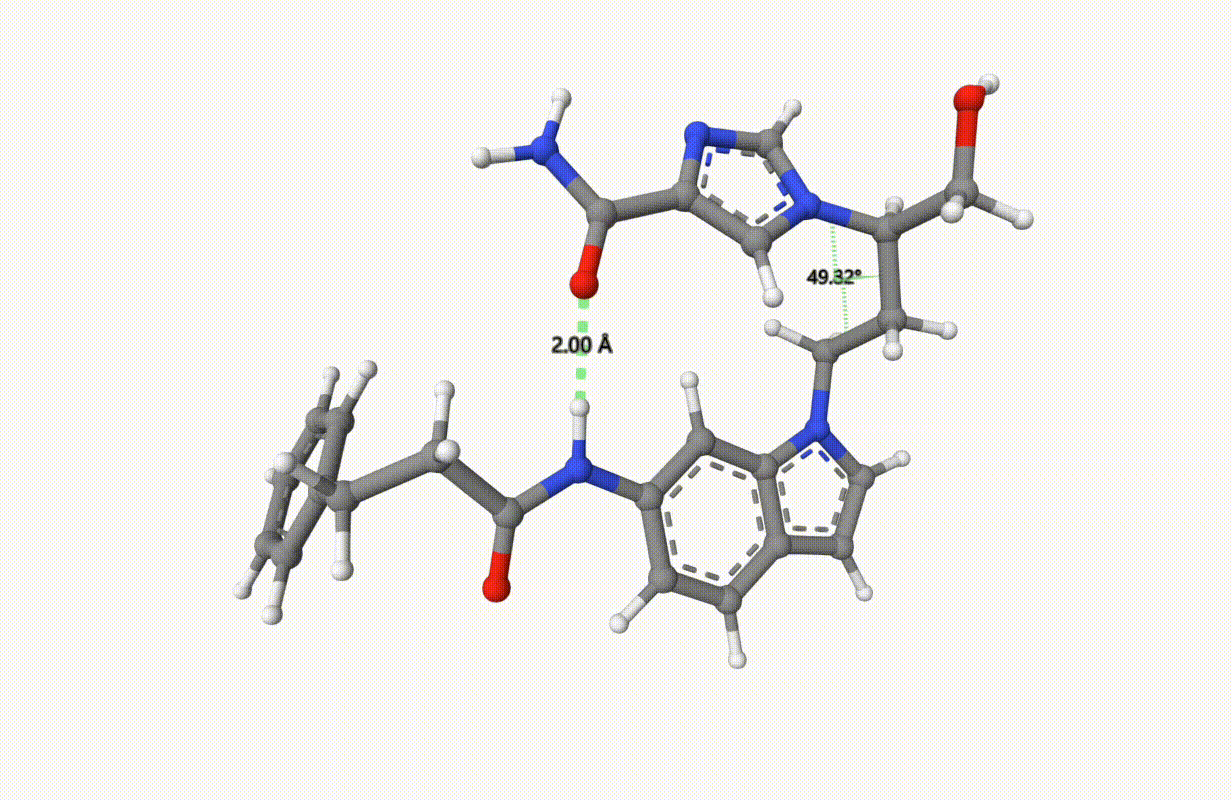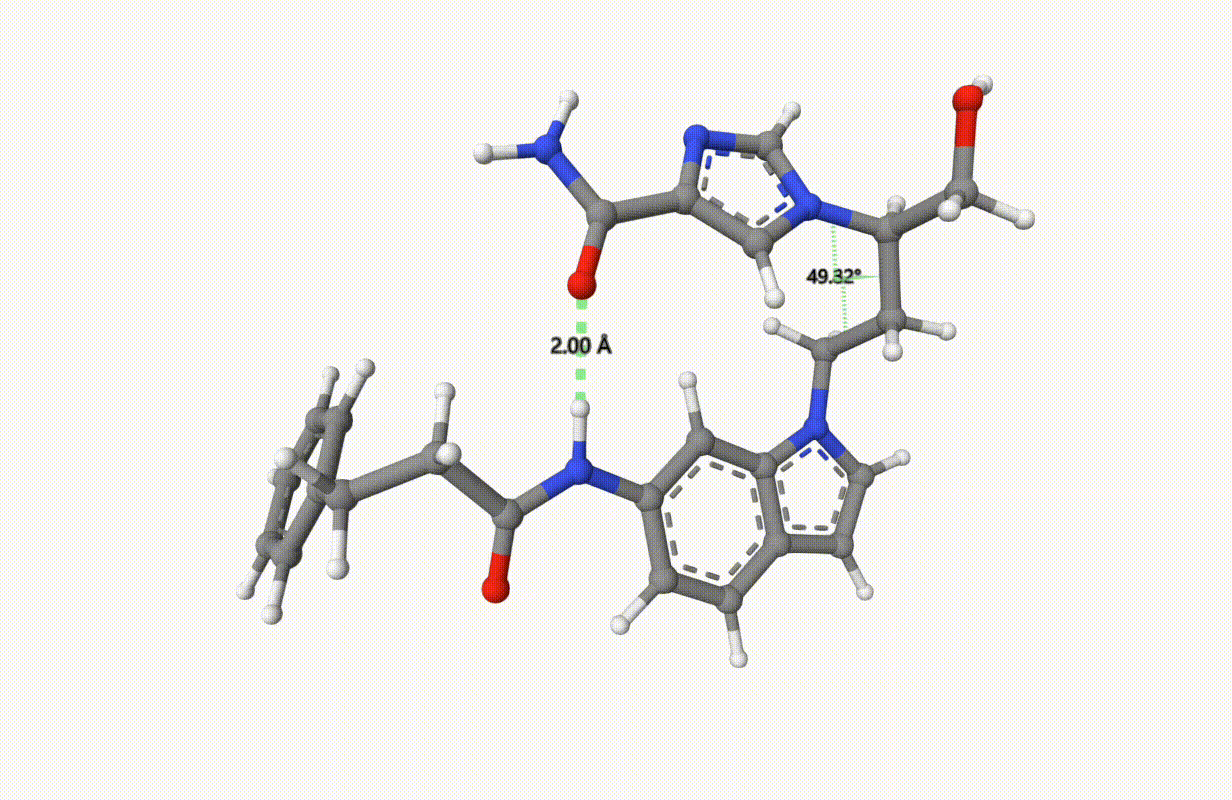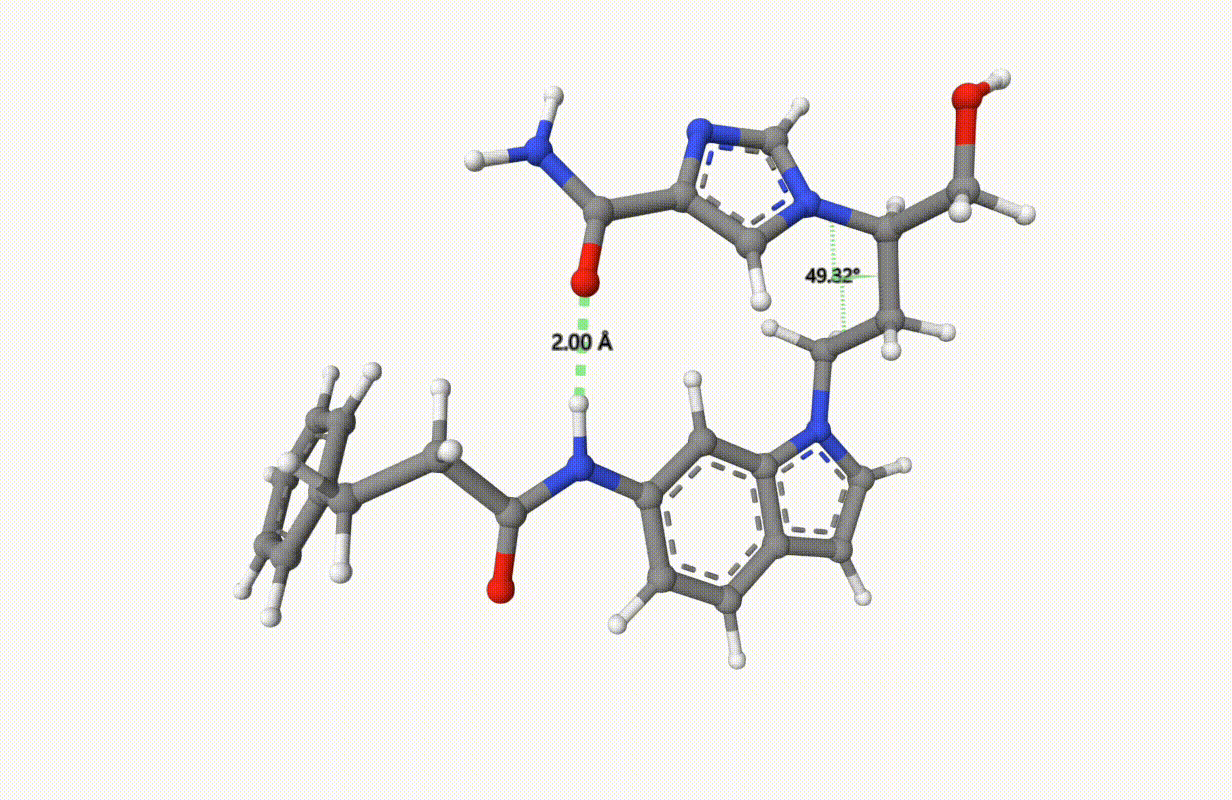
Example: Transition State of SN2 Reaction
Search the transion state of an SN2 reaction with NEB algorithm at B3LYP/def2-SVP level of theory:
1basis # Define basis set.
2 def2-svp
3end
4
5opt
6 type NEB # Type: Min, NEB, Dimer, QST2, TS
7 num_images 10 # The number of images for NEB calculations.
8 neb_k 0.01 # The force constant for NEB calculations.
9end
10
11scf
12 charge -1 # The net charge.
13 spin2p1 1 # 2S+1
14end
15
16xtb
17 chrg -1
18end
19
20mol
21 C -2.25147439 4.89406277 -1.00469981
22 H -1.89481996 3.88525277 -1.00469981
23 H -1.89480154 5.39846096 -0.13104831
24 H -3.32147439 4.89407596 -1.00469981
25 Cl -1.66479756 5.72372709 -2.44173406
26 Cl -2.67350651 4.09697871 0.73250622
27end
28
29mol2
30 C -2.36845504 4.69197207 -0.60149770
31 H -1.76657311 4.00286639 -1.15626927
32 H -1.80200132 5.57659799 -0.39786281
33 H -3.23625780 4.94775799 -1.17280492
34 Cl -1.66479756 5.72372709 -2.44173406
35 Cl -2.86278952 3.94963672 0.91579319
36end
37
38task
39 opt b3lyp
40 # opt xtb # You can also try this.
41end
The reaction path is given in ts-1-opt-traj.xyz:
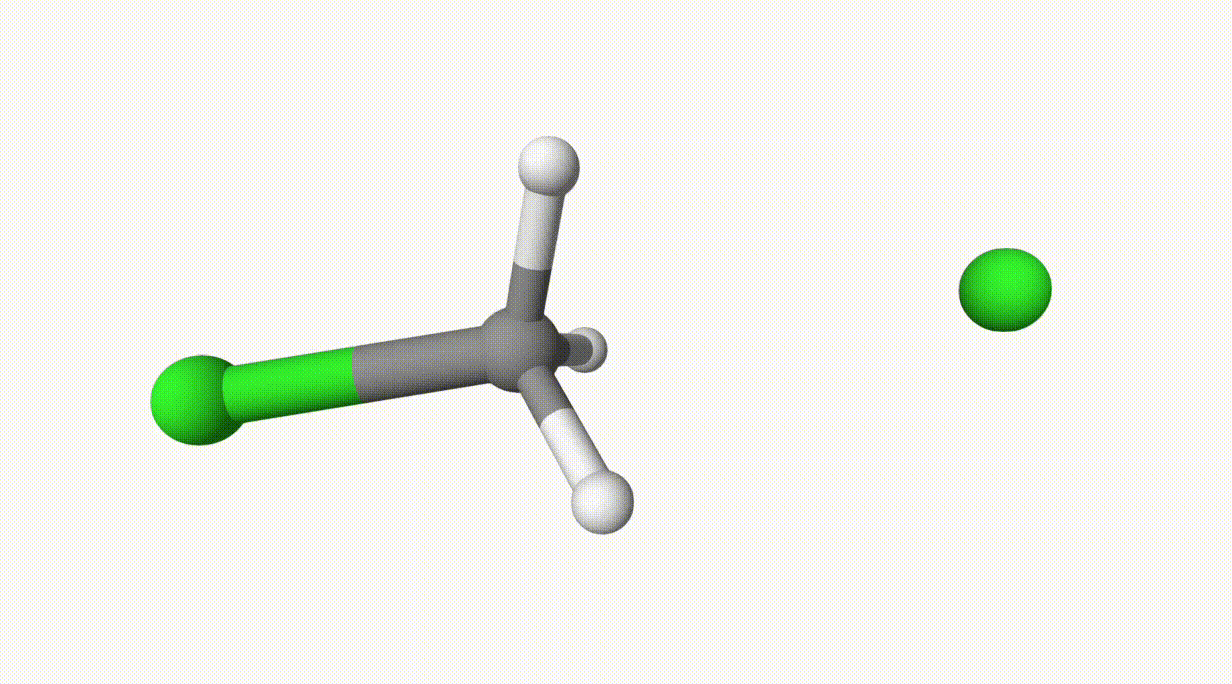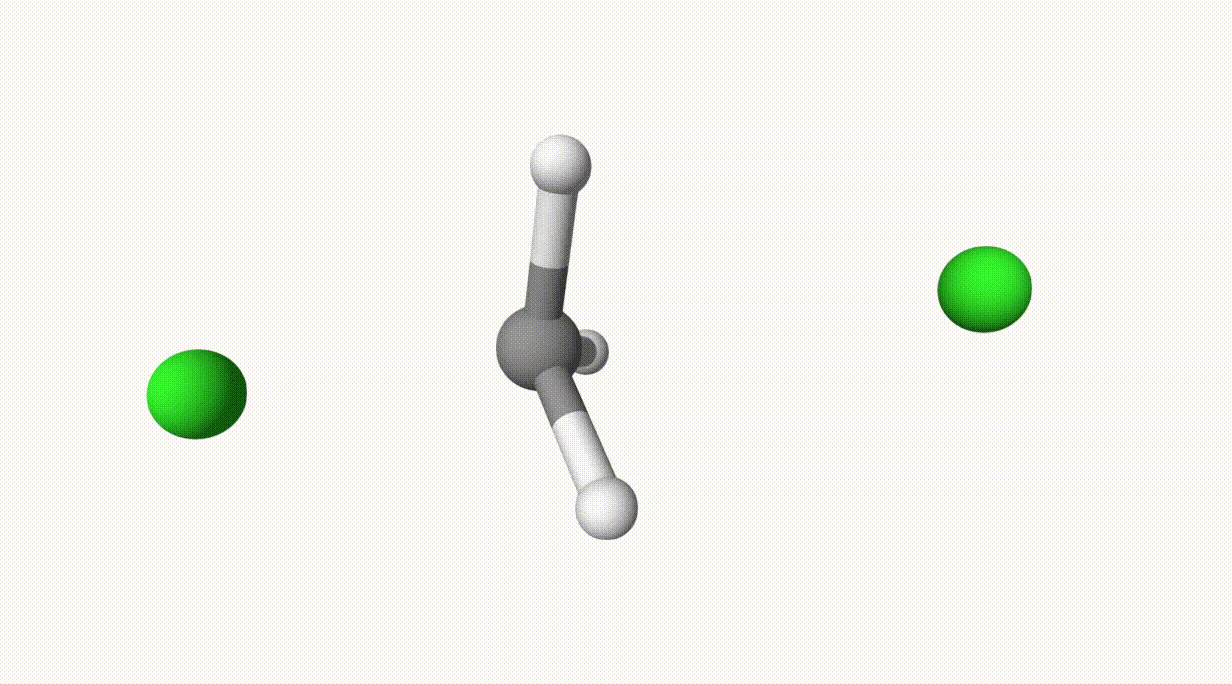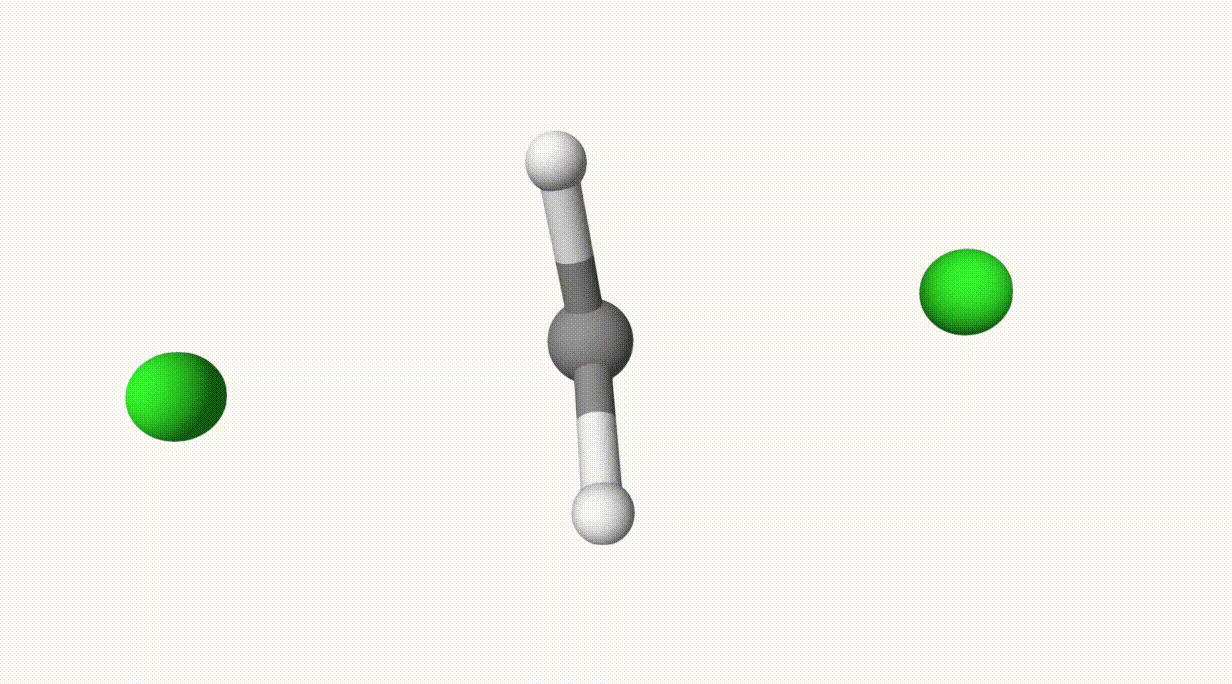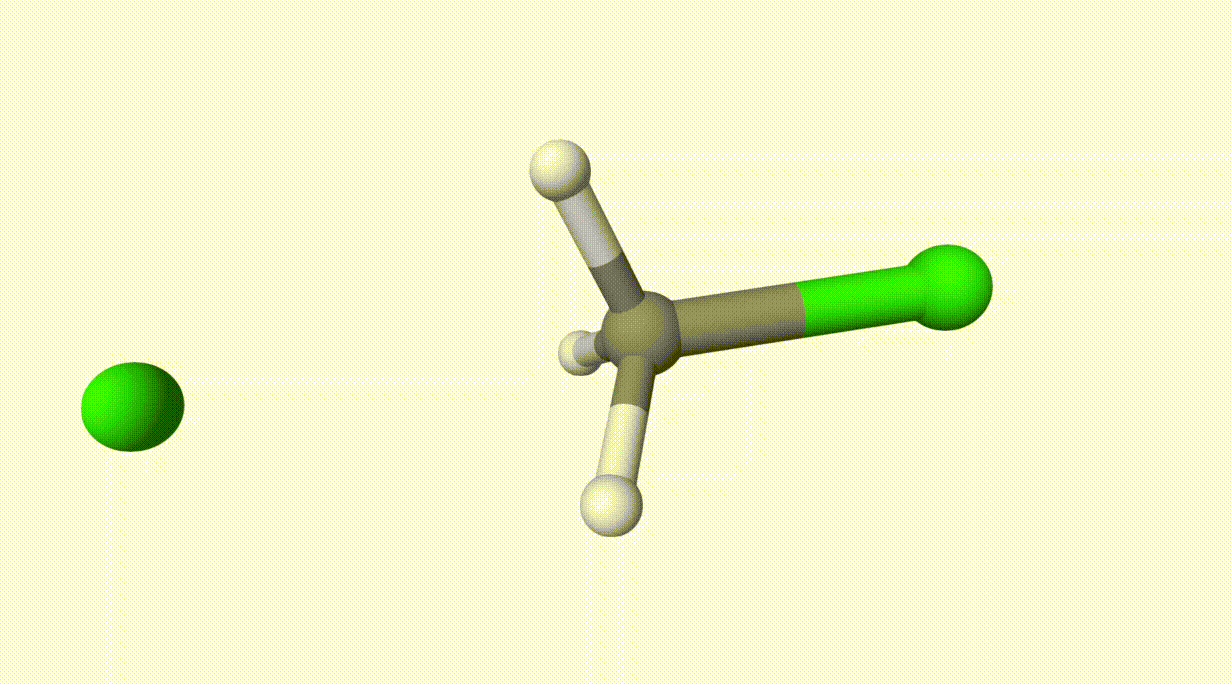
The energies can be found in the output file ts-1.out:
1NEB path updated in "ts-1-opt-traj.xyz":
2----------------------------------------------------
3 # Energy Dist Gtang Gperp
4----------------------------------------------------
5 0 -960.06873748 0.13968 0.00000 0.00000
6 1 -960.06864683 0.11091 0.00029 0.00035
7 2 -960.06738628 0.07888 0.00032 0.00028
8 3 -960.06508634 0.07078 0.00008 0.00030
9 4 -960.06240481 0.09854 -0.00028 0.00036
10 5 -960.05912811 0.18516 -0.00087 0.00032
11 6 -960.05781339 0.21042 -0.00025 0.00051
12 7 -960.06424460 0.26756 -0.00057 0.00032
13 8 -960.06880165 0.00000 0.00000 0.00000
14 9 -960.05738746 0.06067 0.00014 0.00026
15----------------------------------------------------
16
17Geometry optimization step 34:
18 Current energy: -960.05738746
19 Delta Energy: 8.34686E-08; Target: 1.00000E-04; Converged? Yes
20 Max displacement: 2.30167E-04; Target: 4.00000E-03; Converged? Yes
21 RMS displacement: 1.07545E-04; Target: 2.66667E-03; Converged? Yes
22 Max gradient: 5.45965E-04; Target: 1.00000E-03; Converged? Yes
23 RMS gradient: 2.56874E-04; Target: 6.66667E-04; Converged? Yes
24Stationary point has reached.
In the table, structure 0 and 1 are the reactant and product, respectively, and structure 6 is the transition state, which is also given in ts-1-opt.xyz.
You can change b3lyp to xtb to perform the calculation in a much more rapid way.
You can also try type dimer, which is computationally cheaper than NEB but requires reactant and product structures must be of high quality.
Example: Transion State of Decarboxylation Reaction
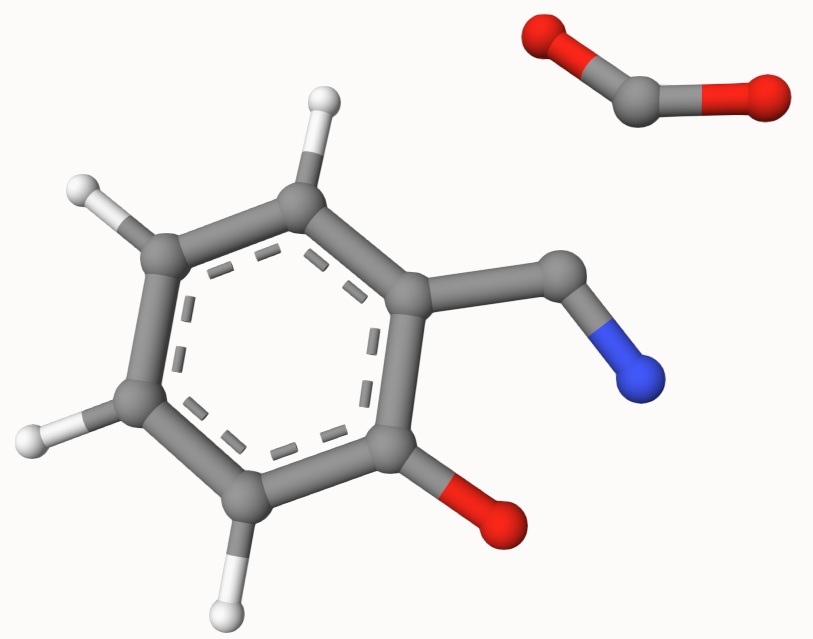
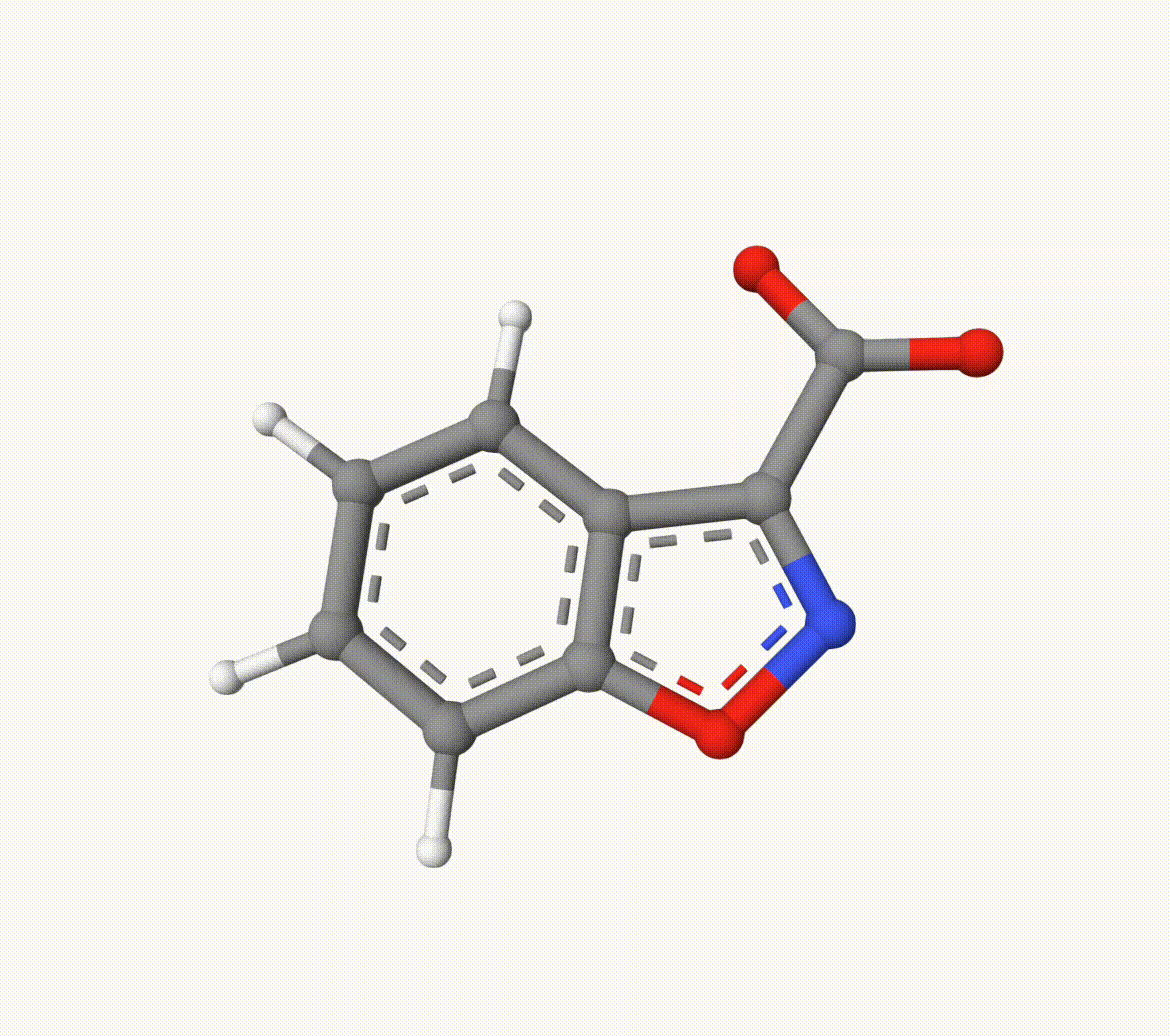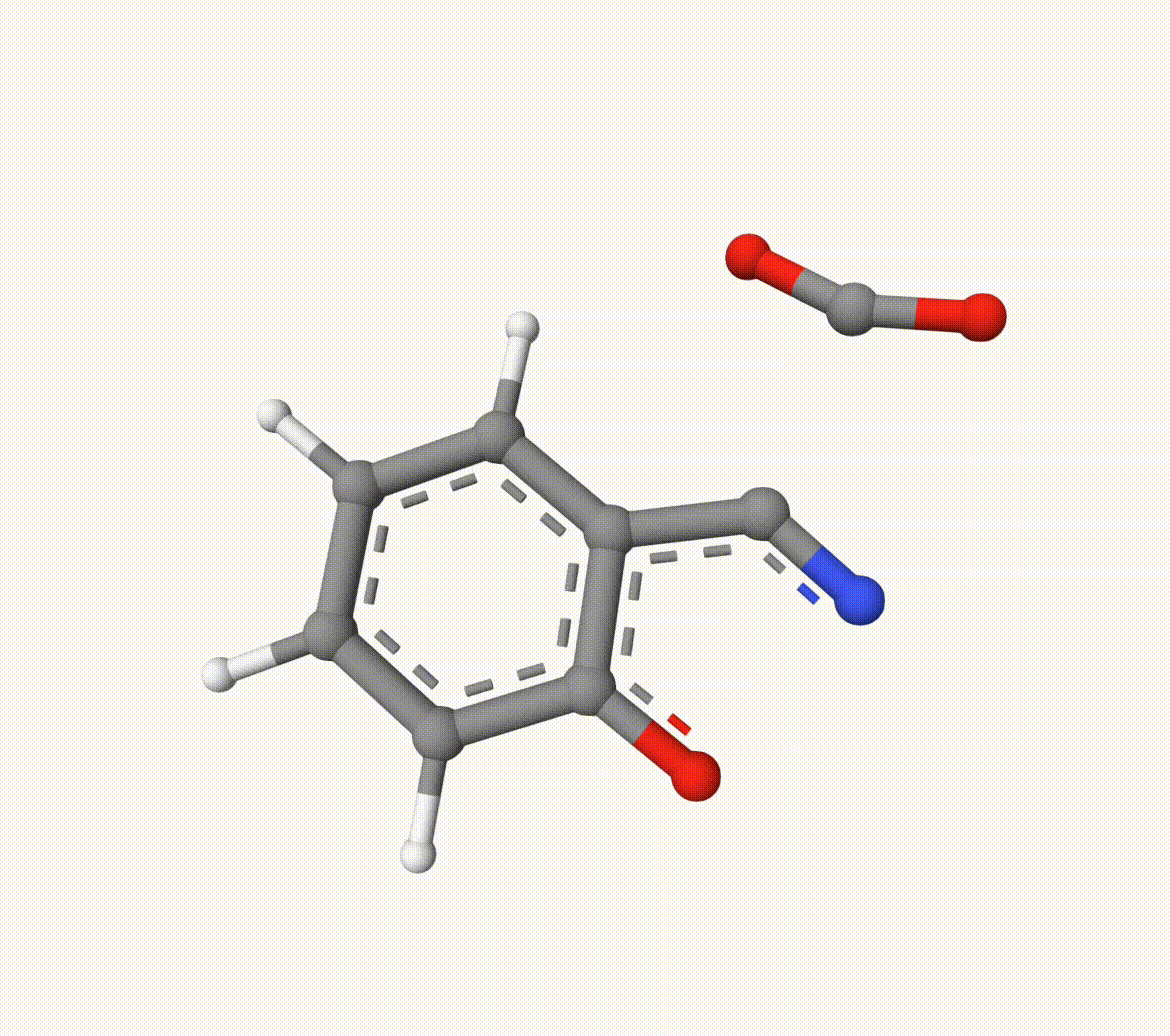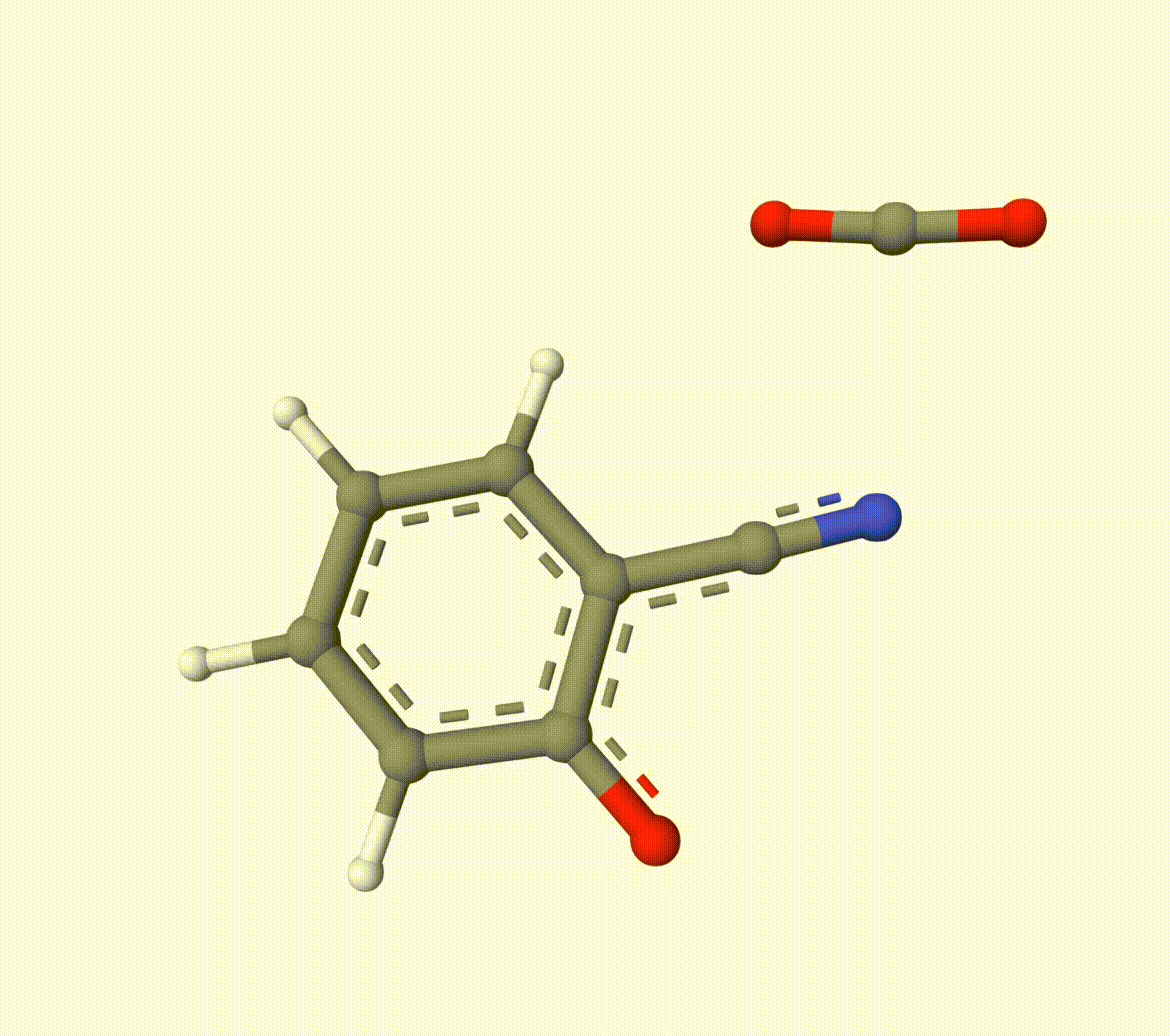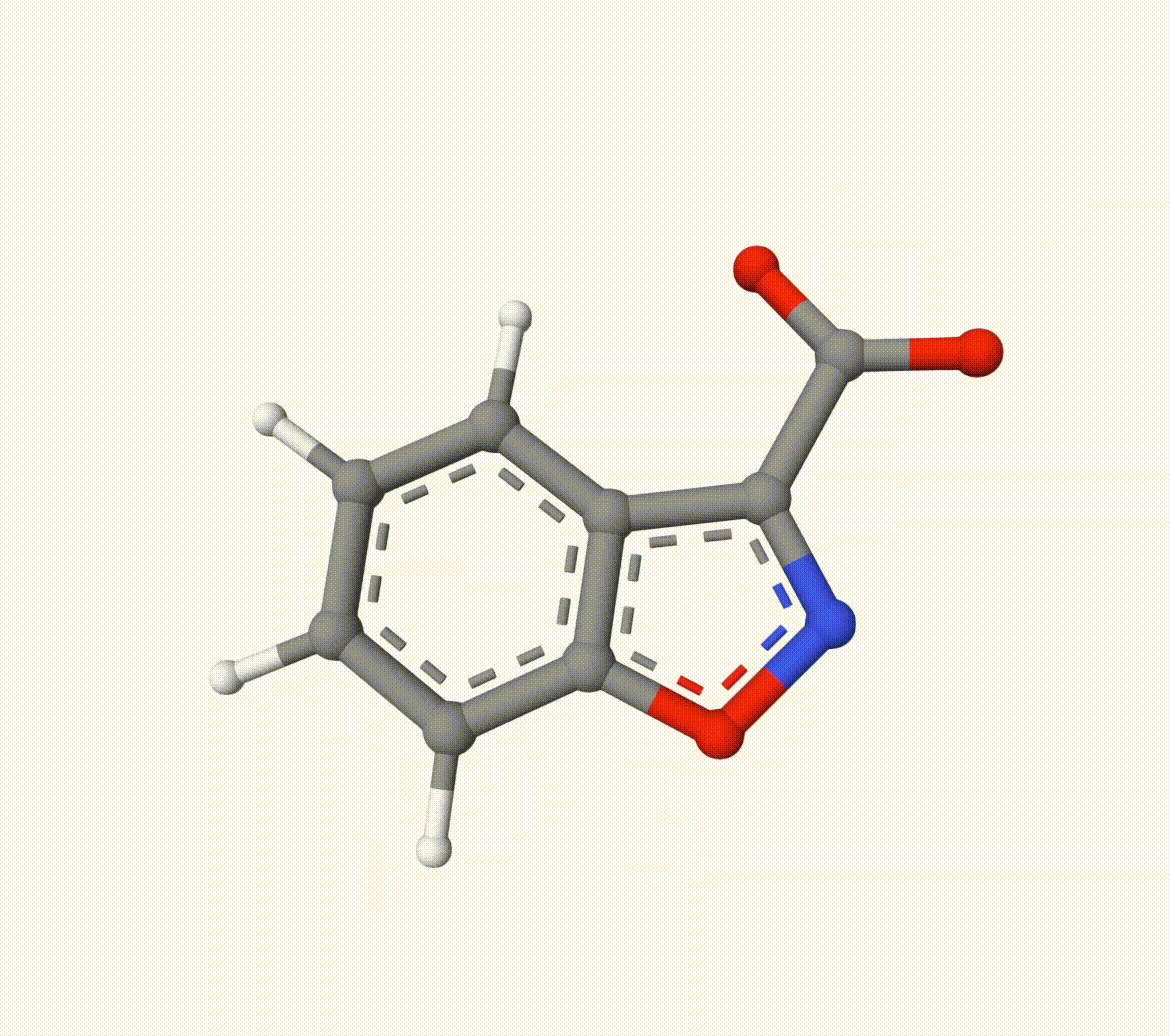
Search the transion state of the following decarboxylation reaction with NEB algorithm at B3LYP/def2-SVP level of theory:
1mol
2 a1.xyz
3end
4
5mol2
6 a2.xyz
7end
8
9scf
10 charge -1
11 spin2p1 1
12end
13
14opt
15 type neb # You can also try "dimer".
16 num_images 15
17 neb_k 0.01
18end
19
20basis
21 def2-svp
22end
23
24task
25 opt b3lyp
26end
116
2E = -587.26844619 Hartree; coordinates in Angstrom.
3 C 0.04106534 -2.18782379 0.01873379
4 C -1.35798345 -2.26546988 0.01269198
5 C -2.17792255 -1.12924287 -0.02333896
6 C -1.53026482 0.10833393 -0.03797859
7 C -0.12071906 0.20403755 -0.02314596
8 C 0.67915283 -0.93908160 -0.00278636
9 C 0.47143440 -3.57161137 0.04441853
10 H -3.26722478 -1.21160460 -0.04107296
11 H -2.12778938 1.02510522 -0.06470926
12 H 0.34489770 1.19455049 -0.03304010
13 H 1.77014421 -0.92296985 -0.00764617
14 O -1.74251880 -3.55390987 0.03659370
15 N -0.56119829 -4.37040148 0.05822977
16 C 1.96372187 -4.05774618 0.02851820
17 O 2.15749685 -5.25776354 0.25204089
18 O 2.75531543 -3.11946977 -0.21744012
116
2E = -34.49382101 Hartree; coordinates in Angstrom.
3 C -0.31300710 -2.30106195 -0.01428127
4 C -1.70090129 -2.13170314 -0.01142600
5 C -2.23889881 -0.83944478 -0.02217974
6 C -1.41495826 0.23848161 -0.03526503
7 C -0.02069520 0.06834558 -0.03813291
8 C 0.51990509 -1.17609015 -0.02785645
9 C 0.29527044 -3.71579140 -0.00257527
10 H -3.29999458 -0.70170793 -0.02007771
11 H -1.83005138 1.22465134 -0.04345765
12 H 0.61932516 0.92576433 -0.04849442
13 H 1.58298567 -1.29755818 -0.03013007
14 O -2.48723821 -3.16847622 0.00114316
15 N 0.74811153 -4.76914425 0.00614076
16 C 2.27925143 -4.13711065 0.09555385
17 O 1.94633923 -5.30045357 0.44105892
18 O 2.61216378 -2.97376825 -0.24995180
Structures in a1.xyz and a2.xyz are shown below. They are put arbitrarily without optimization:
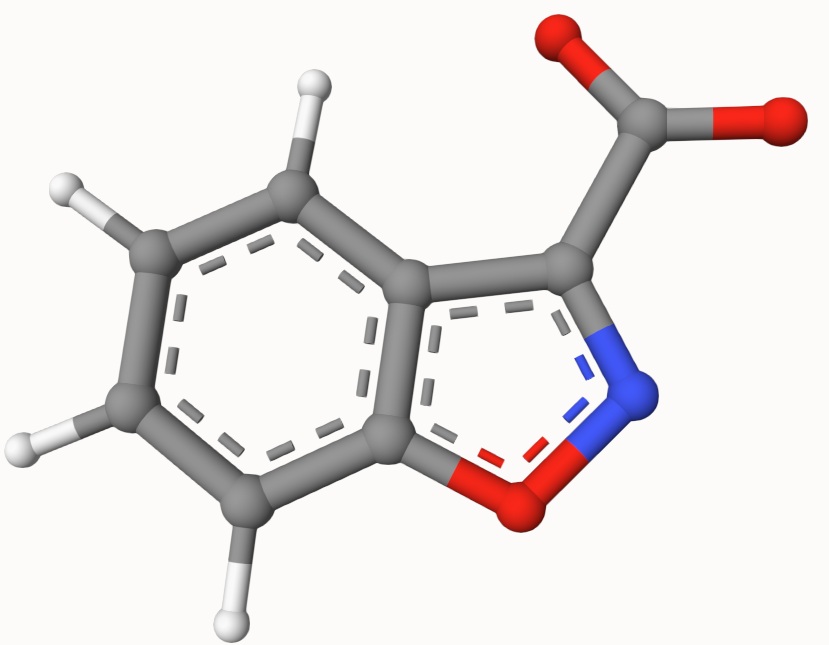
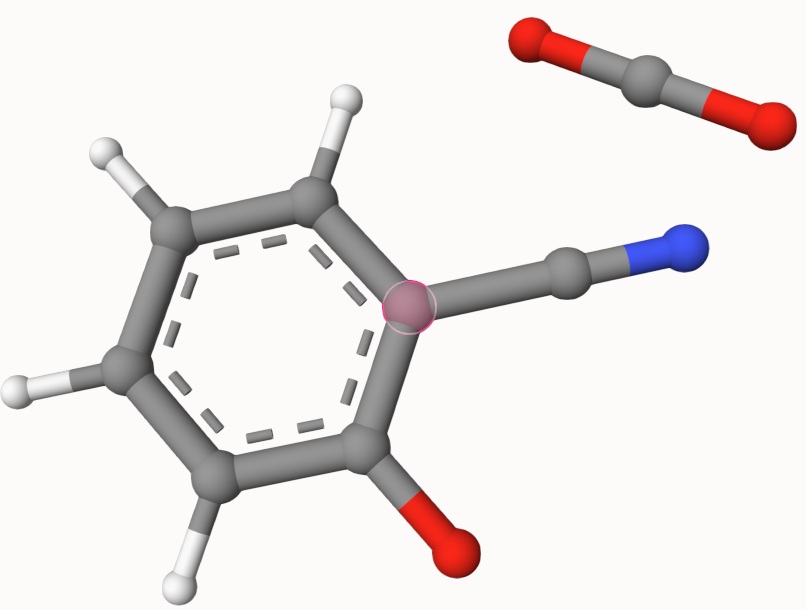
The optimized transtion state ts-2-opt.xyz and path ts-2-opt-traj.xyz are shown below:
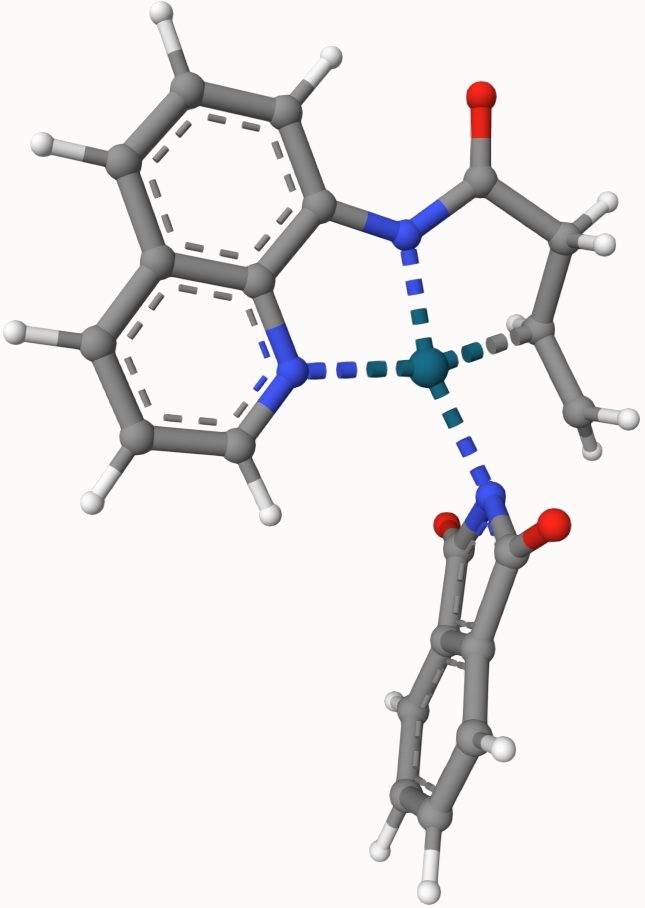
Example: Transion State of Pd(OAc)-Catalyzed Nucleopalladation
Search the transion state of the following Pd(OAc)-catalyzed nucleopalladation with DIMER algorithm at B3LYP/def2-SVP level of theory:
1mol
2 a1.xyz
3end
4
5mol2
6 a2.xyz
7end
8
9scf
10 charge 0
11 spin2p1 1
12end
13
14opt
15 type dimer # You can also use qst2
16end
17
18basis
19 def2-svp
20end
21
22pseudopotential
23 def2-ecp # Since Pd is used, you need to use ECP.
24end
25
26
27task
28 opt b3lyp
29end
143
2element x y z
3 Pd 0.24654 -1.25332 0.40518
4 N 2.21910 -1.24512 -0.02293
5 O 4.02373 -2.55737 -0.59458
6 N 0.71331 0.97790 0.25853
7 C 2.90583 -2.40807 -0.13902
8 C 4.14710 0.14624 -0.66749
9 H 4.76219 -0.71420 -0.85891
10 C 1.86165 3.54336 0.19530
11 H 2.32306 4.52041 0.16979
12 C 2.82401 -0.02669 -0.28507
13 C 0.56127 3.36146 0.56593
14 H -0.05640 4.19293 0.87529
15 C 0.02466 2.06497 0.55234
16 H -1.01171 1.95668 0.81328
17 C 2.10089 -3.60617 0.39103
18 H 2.29784 -3.68424 1.46574
19 H 2.50312 -4.49514 -0.09449
20 C 3.99404 2.53651 -0.49534
21 H 4.44575 3.51746 -0.52470
22 C 0.63638 -3.41684 0.12343
23 C 2.02781 1.13223 -0.06025
24 C 2.63885 2.42073 -0.14140
25 C 4.72289 1.41237 -0.76883
26 H 5.76763 1.48006 -1.03642
27 C -0.35167 -3.32488 1.08900
28 H -1.30757 -3.58022 0.86748
29 H -0.08593 -3.27603 2.15625
30 H 0.32333 -3.63444 -0.90750
31 N -1.81017 -1.32804 0.64184
32 O -2.91585 -2.79869 -0.74520
33 C -2.83467 -1.72049 -0.17802
34 C -2.07940 -0.18984 1.24478
35 O -1.43547 0.22090 2.19254
36 C -3.75316 -0.59348 -0.30558
37 C -3.63015 1.69613 0.35364
38 H -3.18847 2.48978 0.92320
39 C -4.76410 -0.32523 -1.20052
40 H -5.18473 -1.11145 -1.79977
41 C -3.23750 0.40158 0.51050
42 C -4.56763 1.99386 -0.63755
43 H -4.81215 3.01413 -0.85747
44 C -5.15401 0.98013 -1.36177
45 H -5.89024 1.23011 -2.11222
143
2element x y z
3 Pd 0.26512 -0.64711 0.61399
4 N 2.06999 -0.92815 -0.05458
5 O 3.65856 -2.55368 -0.43497
6 N 0.99509 1.45229 0.64166
7 C 2.54673 -2.20957 -0.08507
8 C 4.14972 0.20649 -0.71873
9 H 4.62578 -0.72848 -0.96208
10 C 2.30494 3.86864 0.23224
11 H 2.82051 4.80502 0.07331
12 C 2.84319 0.19559 -0.25035
13 C 1.03545 3.83626 0.74375
14 H 0.50815 4.74282 0.99632
15 C 0.42408 2.59308 0.95757
16 H -0.55466 2.53340 1.41954
17 C 1.49284 -3.20295 0.42350
18 H 1.66001 -3.32238 1.50139
19 H 1.64757 -4.17628 -0.05294
20 C 4.28051 2.61430 -0.57548
21 H 4.82459 3.53688 -0.70941
22 C 0.10935 -2.62297 0.15412
23 C 2.24272 1.45031 0.10134
24 C 2.96211 2.66317 -0.08830
25 C 4.85325 1.40272 -0.86406
26 H 5.87106 1.36062 -1.22258
27 C -1.07037 -3.17687 0.95906
28 H -1.41934 -4.15247 0.60366
29 H -0.83933 -3.23046 2.02995
30 H -0.13100 -2.69707 -0.92522
31 N -2.14266 -2.22101 0.73663
32 O -3.47014 -3.49326 -0.64534
33 C -3.14336 -2.41981 -0.19876
34 C -1.88727 -0.84977 0.97034
35 O -1.25091 -0.41692 1.95149
36 C -3.68247 -1.07281 -0.49566
37 C -3.19644 1.22329 0.10017
38 H -2.60860 1.95263 0.63571
39 C -4.76405 -0.68826 -1.26341
40 H -5.36566 -1.42688 -1.76954
41 C -2.89679 -0.12547 0.17462
42 C -4.28294 1.60635 -0.67387
43 H -4.53744 2.65330 -0.74569
44 C -5.05313 0.66777 -1.34699
45 H -5.89479 0.99766 -1.93734
Structures in a1.xyz and a2.xyz are shown below. They are put arbitrarily without optimization:
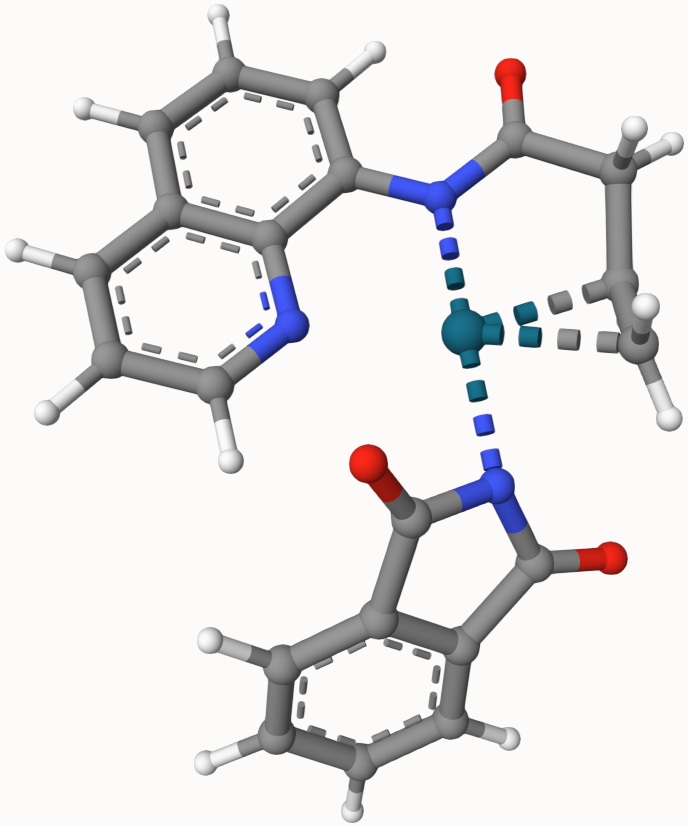
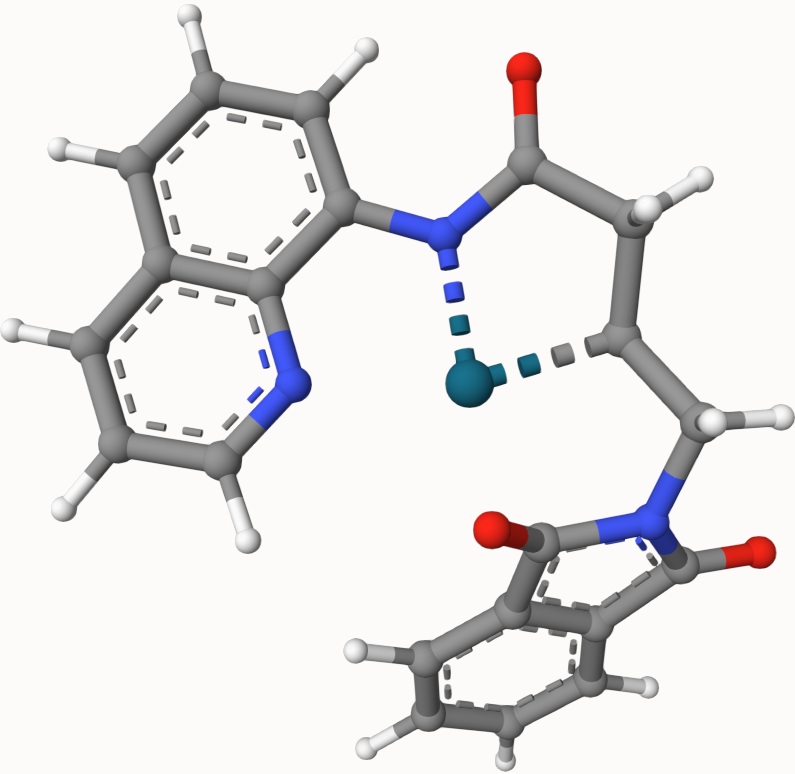
The optimized transtion state is ts-3-opt.xyz, shown below: