Tip
All input files can be downloaded: Files.
Standard Molecular Dynamics Simulations
This tutorial will lead you step by step to do standard molecular dynamics (MD) simulations using Qbics. Assume you have read Geometry Optimization, Density Functional Theory Calculations or Molecular Mechanics Calculations.
In this tutorial, we will only discuss the standard MD simulations, meaning no enhanced sampling algorithms are used. For enhanced sampling MD, please refer to Metadynamics for Calculating Free Energy Surface.
Example: NVT MD for Solvated Protein 5VBM
Here, we will do a NVT MD simulation for the solvated protein 5VBM given in Molecular Mechanics Calculations. The input file is as follows:
1charmm
2 # Here:
3 # toppar_water_ions.str: the parameter file for water and ions.
4 # par_all36m_prot.prm: the parameter file for proteins.
5 # Both are obtained from website: https://mackerell.umaryland.edu/charmm_ff.shtml.
6 parameters toppar_water_ions.str par_all36m_prot.prm
7 topology 5vbm-sol.psf
8 scaling14 1.0
9 rcutoff 12.0
10 rswitch 10.0
11 use_PBC # Add this line to turn on PBC.
12 Lbox 70 70 70 # Define the box size.
13 electrostatic pme # Use PME for electrostatics.
14 PMEk 72 72 72
15end
16
17md
18 type nvt
19 dt 0.001 # 0.001 ps = 1 fs
20 num_steps 10000 # 10000*0.001 = 10 ps
21 temp 300
22 temp_nhc_length 5
23 temp_nhc_tau 0.5
24 output_freq 100 # 0.001*100 = 100 fs
25end
26
27mol
28 5vbm-sol-opt.xyz
29end
30
31task
32 opt charmm
33end
Note that, in mol...end, we use the optimized structure 5vbm-sol-opt.xyz from Geometry Optimization. For biological systems, always use optimized structure as initial structure for MD simulations! Now, we will explain the options in md...end:
Line 18: The type of MD. Here we do canonical ensemble MD, which is
nvt, i.e. constant number of molecules, volume, and temperature.Line 19: The time step size of MD in ps. Usually
0.001is a good choice. However, if there is no hydrogen atom, then0.002or larger can be used.Line 20: The number of steps to do MD. Here we do
10000steps, so the total simulation time is 10000*0.001 = 10 ps. This short time is for pedagogical purpose. In practice, one should do a much longer simulation for product analysis.Line 21: The target temperature in Kelvin.
Line 22,23: The parameters for Nose-Hoover chain algorithm to control temperature. The parameters given here are usually OK.
Line 24: The frequency of output. Here, for every
100steps, the coordinates, gradients, velocities, and energies are output.
After running this calculation, we have the following output:
5vbm-sol-md-traj.xyz: The trajectory.5vbm-sol-md-vel.xyz: The velocities of all atoms for everyoutput_freqsteps.5vbm-sol-md-grad.xyz: The gradients of all atoms for everyoutput_freqsteps.5vbm-sol-md-log.txt: The energies for everyoutput_freqsteps.
For example:
1 # Time Total Kinetic Potential Temp ConstQ Bond Angle Dihedral Improper Coulombic LennardJones Cost
2 ps kcal/mol kcal/mol kcal/mol Kelvin kcal/mol kcal/mol kcal/mol kcal/mol kcal/mol kcal/mol kcal/mol second
3 0 0.00000 -134230.56945744 36085.33154757 -170315.90100501 300.00000000 -134230.56945744 6420.14428661 4495.95531110 1531.31406870 20.78944740 -209097.89905045 26313.79486581 0.129
4 100 0.10000 -133816.54187207 16118.34524766 -149934.88711973 134.00191621 -134016.76556412 7302.41369899 5869.59861893 1666.48807052 75.45725238 -188865.46777364 24016.62295515 6.522
5 200 0.20000 -133606.24161267 16992.08207064 -150598.32368331 141.26583857 -134065.52297681 6679.22007384 5375.41406643 1657.28109955 67.37158807 -186898.20683159 22520.59626219 6.730
6 300 0.30000 -133300.87796459 17301.76543332 -150602.64339791 143.84043065 -134046.30803993 6874.09339659 5371.10977271 1657.72694720 75.62709833 -187077.51167460 22496.31100366 7.048
7 400 0.40000 -132995.61549652 17629.55917628 -150625.17467280 146.56558568 -134041.03818552 6903.10388790 5646.97799910 1681.10341086
8..omitted..
9 9400 9.40000 -100271.14965665 32668.17051219 -132939.32016884 271.59099649 -133856.91550183 10568.00534340 8576.31788333 1692.95053586 88.36593747 -172749.73171403 18884.77179376 7.265
10 9500 9.50000 -99959.15928496 32686.86479432 -132646.02407928 271.74641379 -133845.66058042 10659.61992276 8745.77752012 1703.07775588 96.01140798 -172661.31654362 18810.80580633 7.206
11 9600 9.60000 -99667.33935998 33043.56346211 -132710.90282209 274.71187359 -133850.81824348 10583.87601581 8824.62198457 1700.19011253 89.59345122 -173007.46226334 19098.27782584 7.214
12 9700 9.70000 -99370.72733288 32989.96543886 -132360.69277175 274.26627960 -133846.10700977 10640.12182526 8661.17837744 1693.77491115 96.10476851 -172243.89238876 18792.01968350 7.288
13 9800 9.80000 -99083.18310690 33120.13583885 -132203.31894575 275.34846780 -133847.97704020 10723.48783126 8787.10704944 1698.42095679 98.19639577 -172644.29859784 19133.76736774 7.495
14 9900 9.90000 -98790.21971731 33390.37977009 -132180.59948741 277.59517514 -133840.46539948 10862.67638260 8744.32303648 1685.93747509 97.87623972 -172406.04428515 18834.63161277 7.240
1510000 10.00000 -98511.94567150 33524.11389736 -132036.05956887 278.70699084 -133842.52309227 10850.32141702 8657.09756469 1697.76177767 90.53706975 -172324.05730779 18992.27985877 7.197
We can see that the time, total energy, kinetic energy, potential energy, temperature and other components are output. Here, ConstQ is a conserved quantity for NVT simulation. If it strongly fluctuates during simulaiton, the MD calculation may be problematic. Also, the NVT MD is tried to keep the temperature constant at 300 K, so the temperature should not fluctuate too much. At the beginning, the temperature is around 134 K, which is not good. This means that the system has NOT been equillibrated yet. After 9 ps, the temperature is stabilized around 300 K. In practice, one should do a longer MD simulation to ensure the system is equilibrated, and then do production MD simulation.
We can visualize the trajectory 5vbm-sol-md-traj.xyz in Qbics-MolStar. In Qbics, we can load topology 5vbm-sol.psf into Model and trajectory 5vbm-sol-md-traj.xyz into Coordinates, then click Apply to load the trajectory. Now click the icon in the left-up corner, and click Start. Now you can visualize the trajectory.
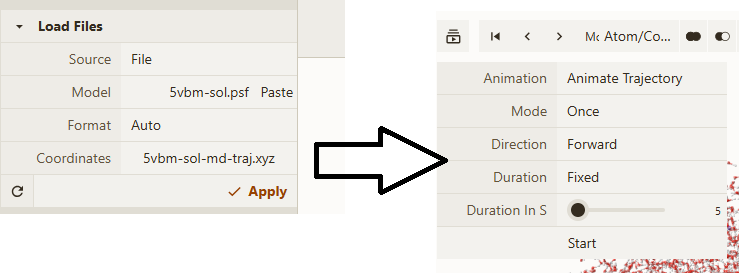

Now you can do various analyses on energies, trajectories, gradients, and velocities. For example, calculating diffusion coefficient, conformation change, etc.
Example: NVE MD for Bullvalene
The MD using CHARMM force is sometimes called classic MD (CMD). Thus, the MD using quantum chemical force (like DFT, xTB) is called ab initio MD (AIMD). We can use AIMD to study dynamic effects in chemical reactions.
Below, we will do a NVE MD simulation for bullvalene. We use NVE since in this case, the system does not exchange energy with environment, thus it is an isolated system, the energy will relax within its internal degrees of freedom. We use the following input:
1xtb
2 gbsa CHCl3
3end
4
5md
6 type nve
7 dt 0.001 # 0.001 ps = 1 fs
8 num_steps 100000 # 10000*0.001 = 10 ps
9 temp 500
10 output_freq 10 # 0.01*100 = 0.01 ps
11end
12
13mol
14 C -0.62880 1.05561 1.34009
15 H -0.83035 1.51659 2.30210
16 C 0.13239 1.54079 0.47336
17 H 0.64136 2.33325 0.99298
18 C -0.88383 -0.82066 1.33606
19 H -1.37100 -1.05330 2.29317
20 C 0.79049 -0.51831 1.33499
21 H 1.19125 -0.81191 2.27882
22 C 0.49419 1.22452 -0.75522
23 H 1.22559 1.79534 -1.30152
24 C -1.52626 -0.95189 0.16256
25 H -2.43289 -1.51994 0.39341
26 C 1.56332 -0.93823 0.11905
27 H 2.41452 -1.49844 0.38314
28 C -0.08122 0.08636 -1.54946
29 H -0.10037 0.16642 -2.63781
30 C -1.23434 -0.67040 -1.08946
31 H -1.91475 -1.01441 -1.86813
32 C 1.16111 -0.58641 -1.09467
33 H 1.84928 -0.87572 -1.88796
34end
35
36task
37 md xtb
38end
Here, we use xtb (with implicit solvent gbsa CHCl3) since it is much faster than DFT. Of course, you can change xtb to b3lyp to do a DFT AIMD. From md...end, we can see that we do an NVE MD for 10 ps, with a step size of 1 fs and initial temperature of 500 K.
The initial structure is a transition state. Such AIMD is sometimes called downhill dynamics. If we start the AIMD with a structure minimum (reactant), this is called uphill dynamics.
After this downhill dynamic simulaiton, we can use Qbics-MolStar to visualize the trajectory:
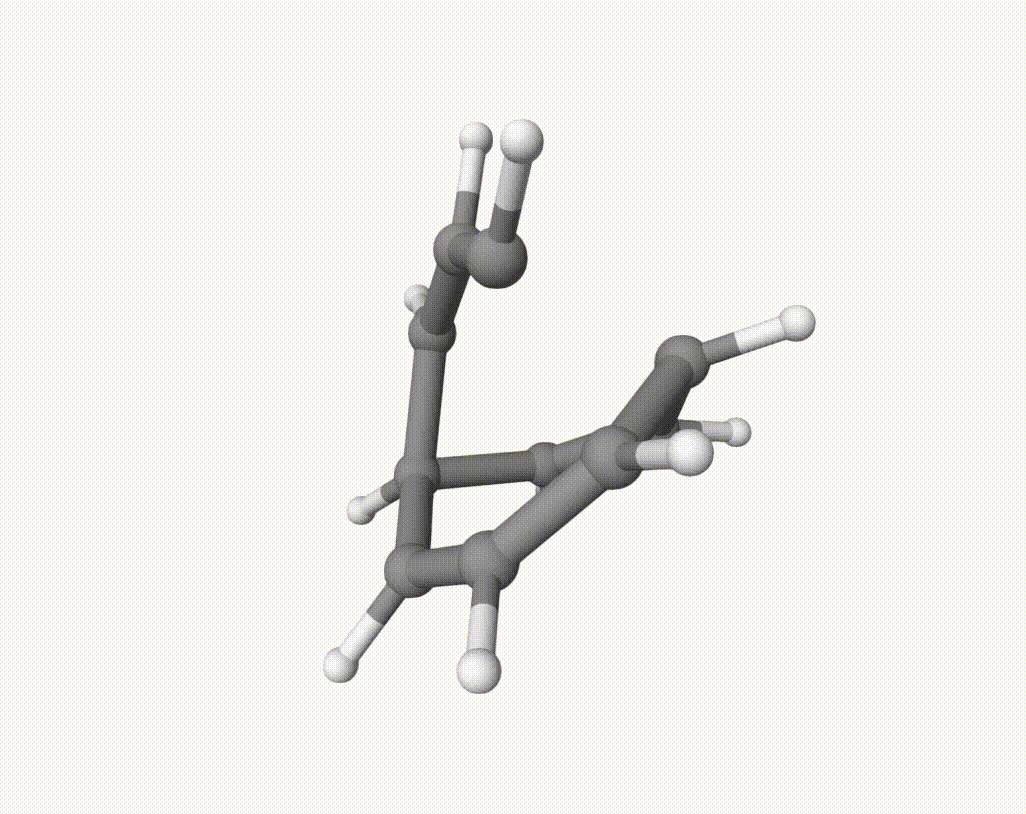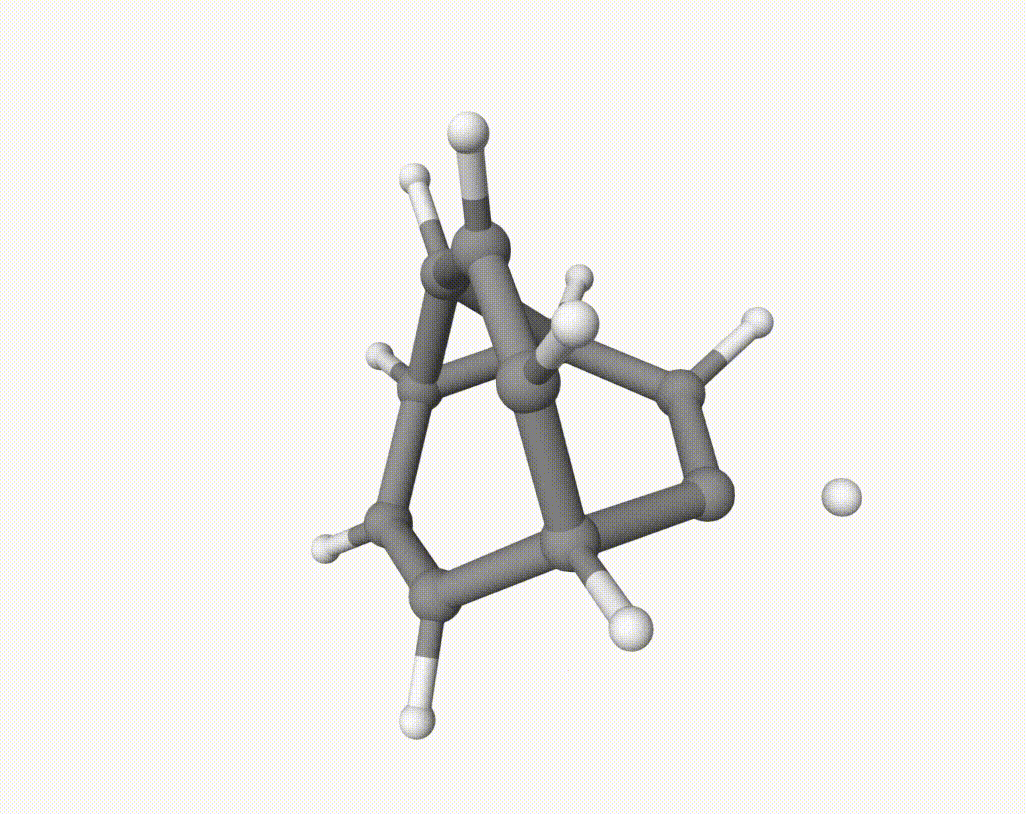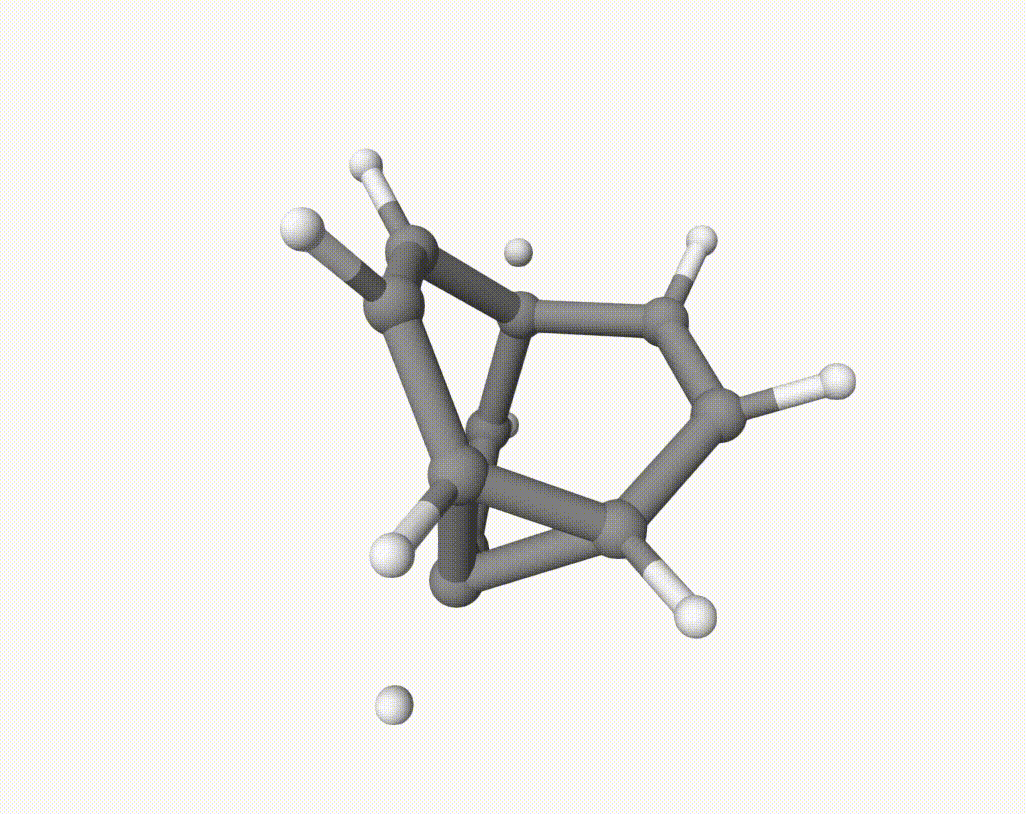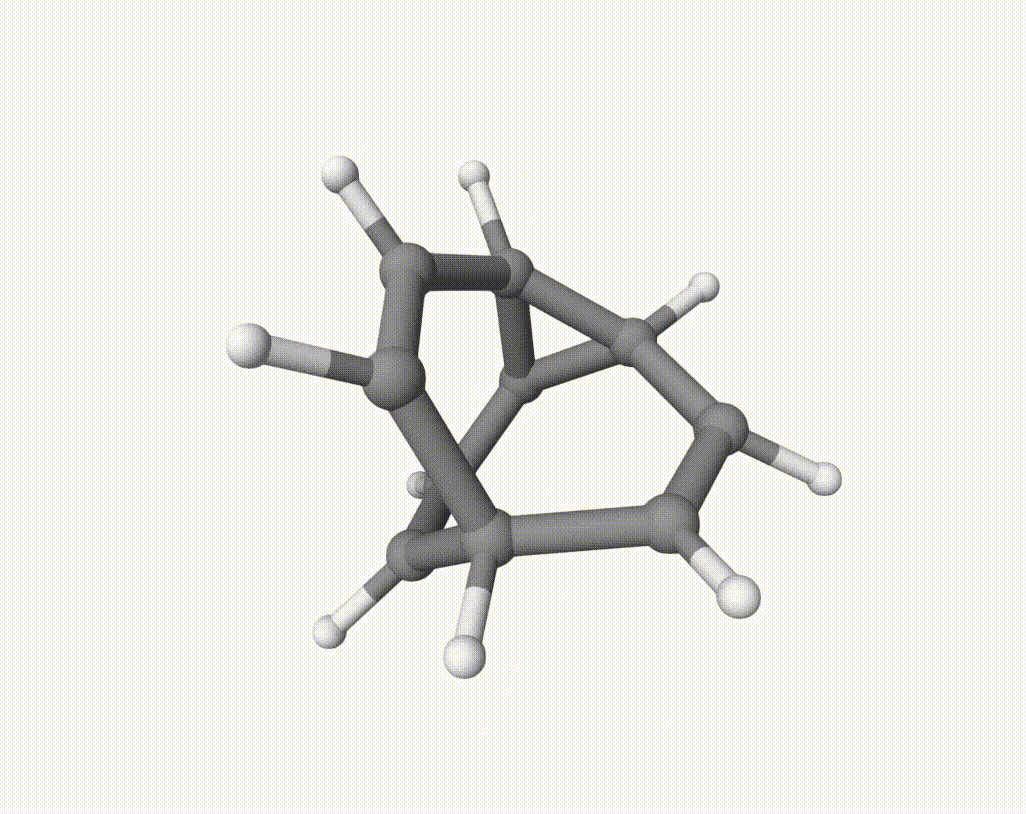
There are several isomerization reactions occurred. For example, during 2.00 - 3.37 ps:
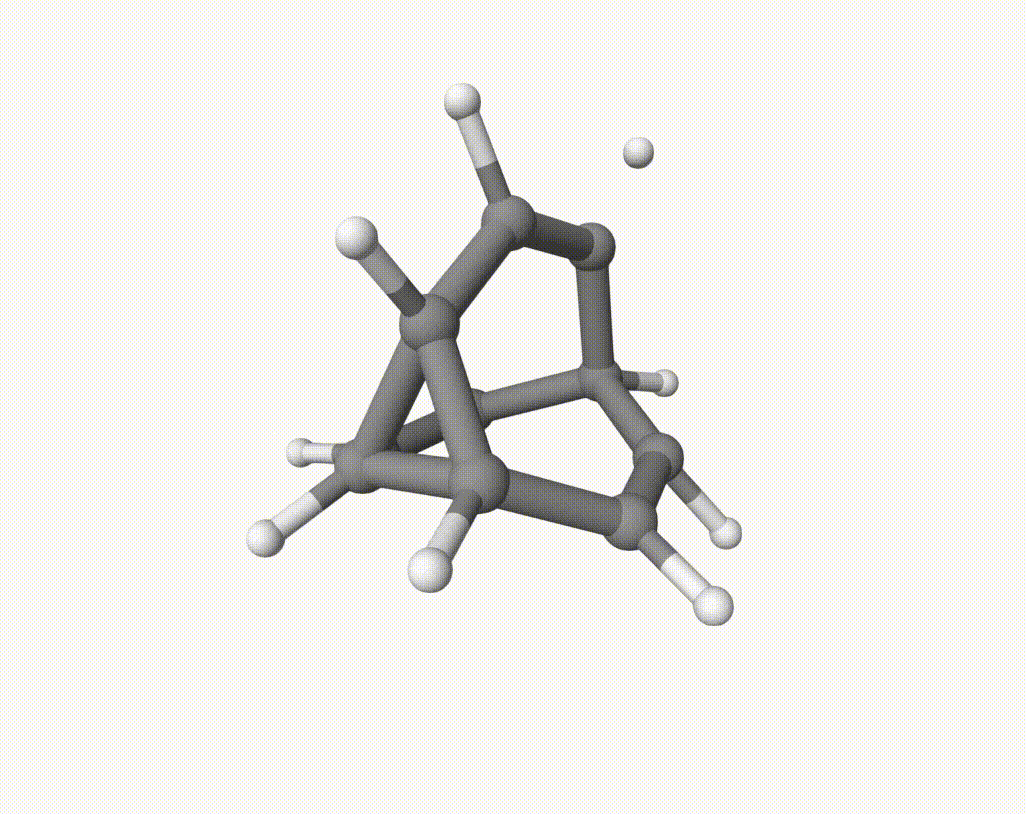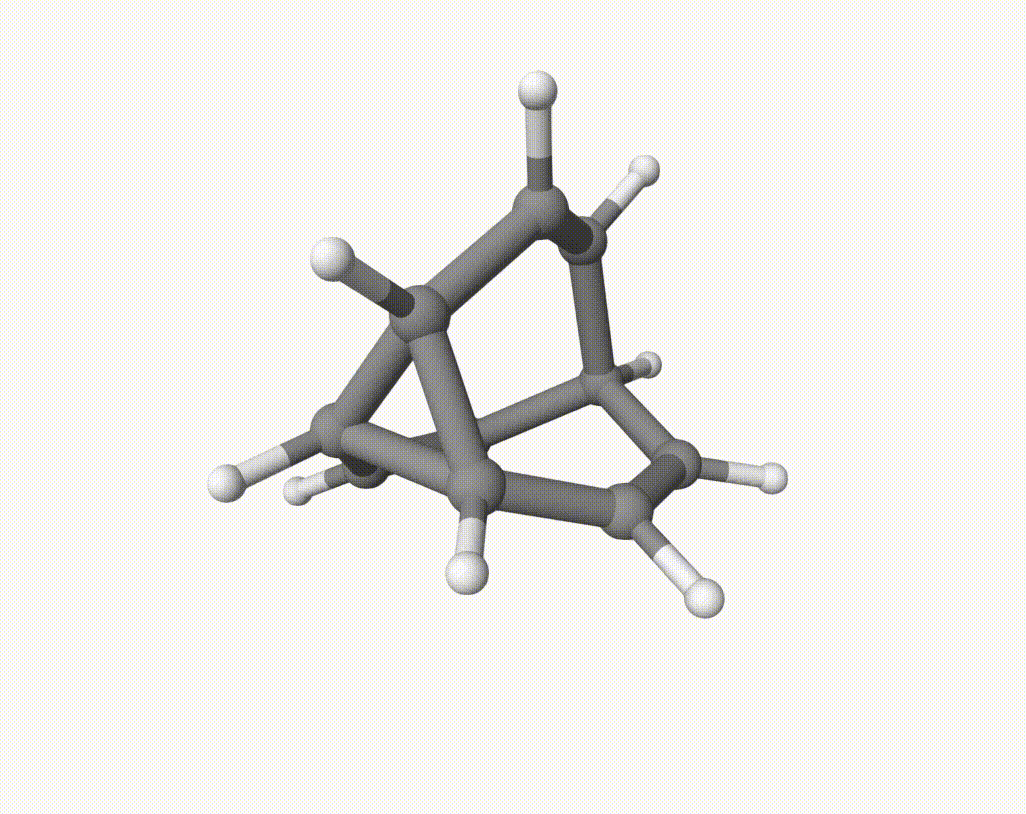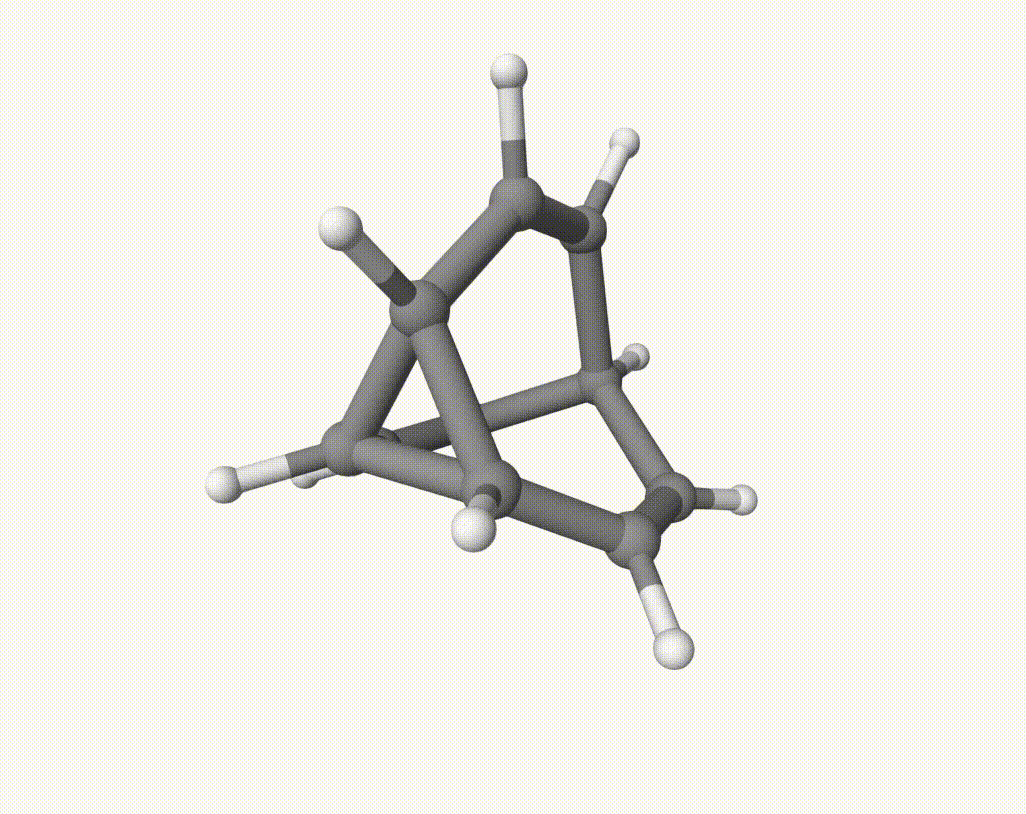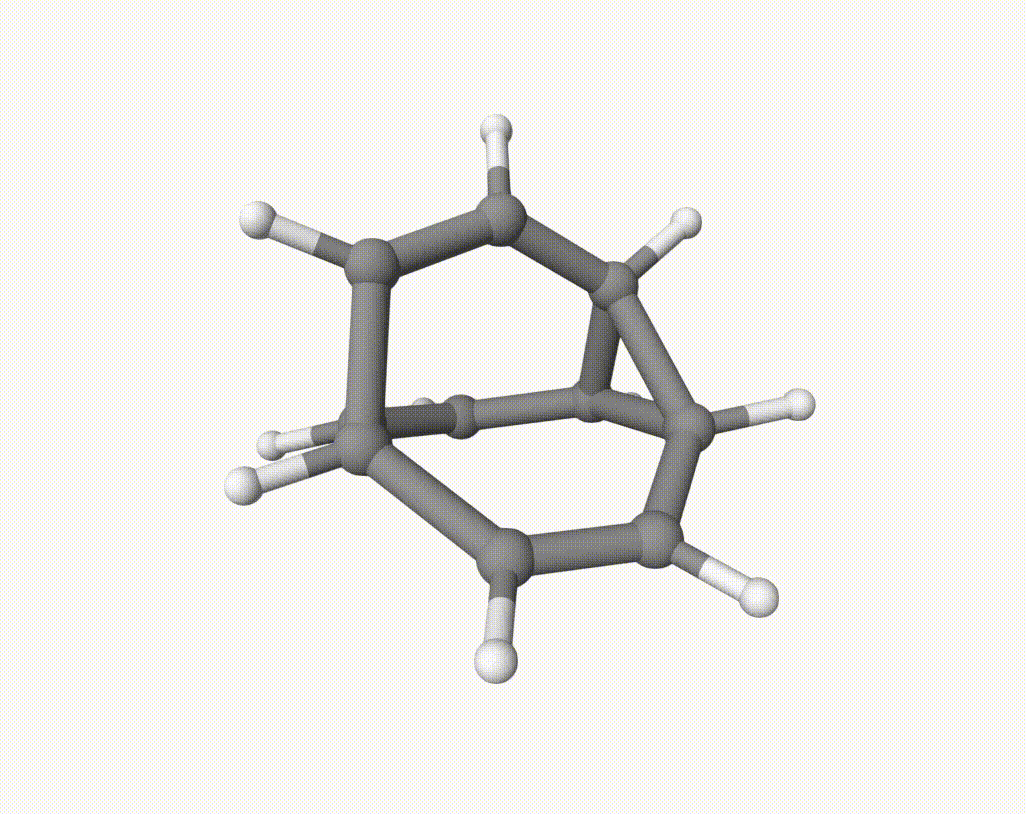
which corresponds to a Cope rearrangement:
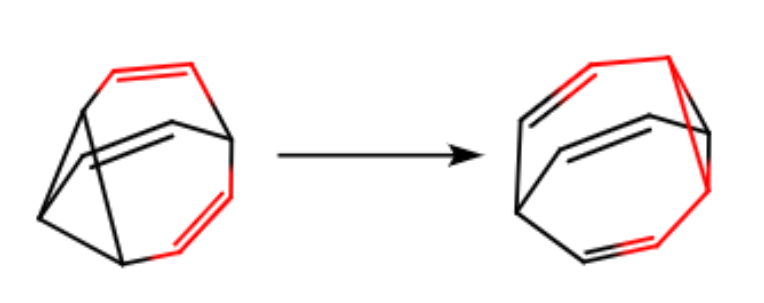
With this trajectory, you can study the reaction path or rate.