Tip
All input files can be downloaded: Files.
qmmm
This option controls the quantum mechanics/molecular mechanics (QM/MM) calculation details.
Options
- qm_region
Value
Atom range
Default
None
Define the atoms in the QM region.
1qmmm 2 qm_region 1-14 16 23 45-48 3end
In this input, atom 1,2,3,4,5,6,7,8,9,10,11,12,13,14,16,23,45,46,47,48 will be treated with QM methods, and others will treated with MM methods.
- link
Value
imommIntegrated molecular-orbital molecular mechanics (IMOMM) mehtod.phoPorjected hybrid orbital (PHO) method.Default
imommDefine the method to treat of the boundary covalent bonds between QM and MM regions. In Qbics, there are two methods:
imommandpho. Theimommmethod will add a hydrogen atom to saturate the boundary valency; thephomethod will not add any hydrogen atom, but will use a special set of projected hybrid orbital to treat the boundary covalent bonds. Both methods are implmented in a black-box manner, and users do not need to worry about the details of the methods. Their details can be found in the references below.In Qbics, “covalent bonds” are defined in the PSF file (given by
topologyincharmm). In the QM region, all bonds are possible to break and form, while in the MM region, covalent bonds defined in the PSF file NEVER break and form. The boundary covalent bonds between QM and MM regions are NOT allowed to break. In the figure below, the QM region is defined by the green circle. For the left case, since no covalent bonds are defined between QM and MM regions, thelinkoption is ignored (no matter what value given). For the right case, there are 3 covalent bond between QM and MM regions (red bonds), and thelinkoption is used to treat these bonds.In Qbics, only sp3 carbon can be used at the boundary (It must be included in the QM region).
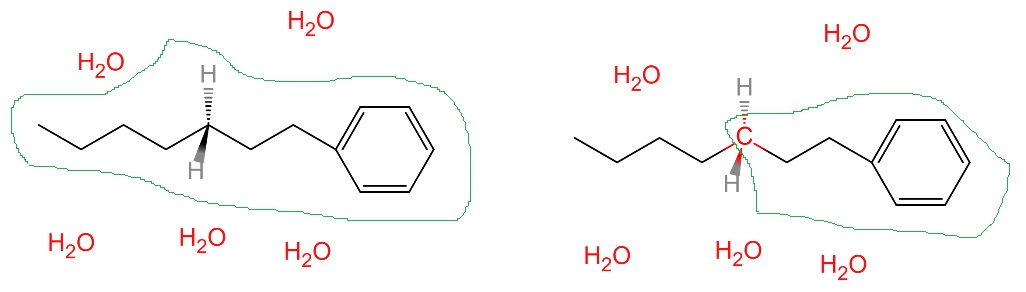
Both methods can treat the boundary covalent bonds. The
phomethod is more accurate for ground states; butimommmethod can be combined with TSO- or BLE-SCF (see scf) to calculate the excited and diabatic states.
Theoretical Background
Quantum Mechanics / Molecular Mechanics (QM/MM) is a multiscale computational strategy that combines the accuracy of quantum-mechanical (QM) methods for a chemically active region with the speed of molecular-mechanical (MM) force fields for the surrounding environment. In Qbics, the QM region can be treated with DFT, xTB, or NDDO methods, while the MM region is typically described by CHARMM or AMBER force fields. This approach allows for efficienzt simulations of large chemical systems.
To do QM/MM simulations, first one have to prepare force field parameters and topology for the whole system, then use qm_region to define the QM region. If there ARE covalent bonds between atoms in QM and MM region, the boundary atoms must be sp3 carbon (i.e., carbon atom linking to 4 atoms). Other atoms are NOT supported to be boundary. This is a reasonable restriction since in these cases the molecular orbitals can be significantly delocalized over QM and MM region, those bonds should not be cut. Based on link option, Qbics will call the corresponding method to treat boundary atoms.
There are two methods for link: imomm and pho. The IMOMM method adds a hydrogen atom to saturate the boundary valency, while the PHO method uses a special set of projected hybrid orbitals to treat these bonds without adding hydrogen atoms. Both methods are implemented in a black-box manner, allowing users to focus on their calculations without needing to understand the underlying details. The main points are:
IMOMM: will add additional hydrogen atoms. Can be used for both ground and excited states.*
PHO: will NOT add any additional atoms. Can only be used for ground states.
Input Examples
Example: Solvated Organic Anion
In this example, we will do a QM/MM calculation for a water microdroplet of an organic anion. The input is given below:
1charmm
2 parameters toppar_water_ions.str 92v-1-naive.prm
3 topology 92v-solvate.psf
4 scaling14 1.0
5 rcutoff 12.0
6 rswitch 100.0 # rswitch>rcutoff means no cutoff is applied.
7end
8
9xtb
10 chrg -1 # The charge in QM region!
11 uhf 0 # The number of unpaired electrons in QM region!
12end
13
14qmmm
15 qm_region 1-39
16end
17
18opt
19 max_step 2000
20end
21
22mol
23 92v-solvate.pdb
24end
25
26task
27 opt xtb/charmm
28end
In qmmmm-1a.inp, we have given the charmm and xtb blocks to define the force field parameters and the QM method. The mol block is used to load the cooridnates; The topology is defined in the PSF file (topology in charmm); the parameters are defined in parameters in charmm. The task block is used to run the optimization with QM/MM method. Note that this is a gas phase optimization, so no cutoff is applied (rswitch is set to be larger than rcutoff).
Another important point is that in xtb, the charge and number of unpaired electrons are defined only for in the QM region!
After optimization, the structure is stored in qmmm-1a-opt.xyz, shown below:
We can use it as the structure to do a B3LYP-D3BJ/def2-svp single point energy:
1charmm
2 parameters toppar_water_ions.str 92v-1-naive.prm
3 topology 92v-solvate.psf
4 scaling14 1.0
5 rcutoff 12.0
6 rswitch 100.0 # rswitch>rcutoff means no cutoff is applied.
7end
8
9xtb
10 chrg -1 # The charge in QM region!
11 uhf 0 # The number of unpaired electrons in QM region!
12end
13
14basis
15 def2-svp
16end
17
18scf
19 charge -1 # The charge in QM region!
20 spin2p1 1 # The spin multiplicity in QM region!
21end
22
23grimmedisp
24 type bj
25end
26
27qmmm
28 qm_region 1-39
29end
30
31mol
32 qmmm-1a-opt.xyz
33end
34
35task
36 energy b3lyp/charmm
37end
At the end of qmmm-1b.out, you can find this:
1Total QM/MM energy
2==================
3Polarized QM energy: -1608.98518142 Hartree
4Whole system MM energy: -90.52845586 Hartree
5QM/MM energy correction: 16.29439183 Hartree
6------------------------------------------------
7QM/MM energy: -1683.21924545 Hartree
8
9Final total energy: -1683.21924545 Hartree
QM/MM energy is the total energy.
In all above cases, you can change energy or opt to md to do a QM/MM MD simulation.
Example: Residues in Solvated Protein
In this example, we will show the QM/MM energy calculation of two interacting residues in a protein. Assume we have already prepared a solvated protein, shown below:

We will choose two residues on a beta-sheet region, i.e. ILE44 and HSD68, which are far away from each other on sequence but are quite close in 3D space. We also include some of their neighboring atoms. The atom indices are shown below:
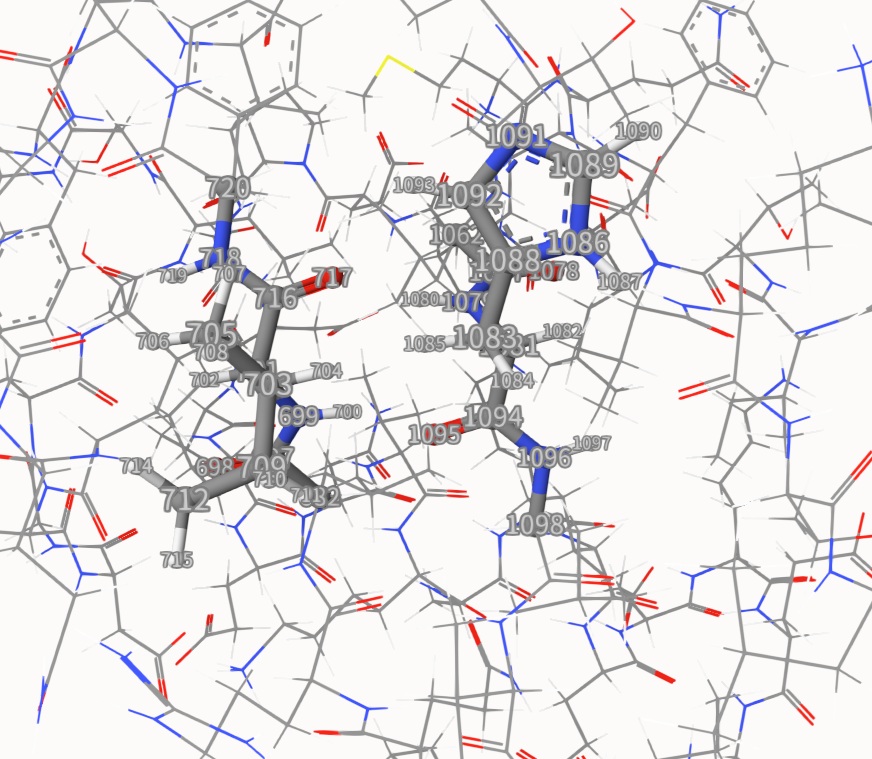
The QM region is cut at 4 sp3 carbon atoms, also shown below. All boundary carbon atoms link to 3 MM atoms:

Thus, the input file is:
1charmm
2 parameters toppar_water_ions.str par_all36m_prot.prm
3 topology sys.psf
4 scaling14 1.0
5 rcutoff 12.0
6 rswitch 10.0
7 use_PBC # Add this line to turn on PBC.
8 Lbox 64 64 64 # Define the box size.
9 electrostatic cutoff # Use cutoff or QM/MM.
10end
11
12basis
13 def2-svp
14end
15
16scf
17 charge 0
18 spin2p1 1
19end
20
21qmmm
22 qm_region 682 697-720 1062 1077-1098
23 link imomm # or change to: pho
24end
25
26mol
27 sys.xyz
28end
29
30task
31 energy b3lyp/charmm
32end
The above input should be self-explanatory, if you have already known how to use Qbics to do QM and MM calculations. The only point is that currently, if the periodic boundary condition (PBC) is turned on, it is recommended to use cutoff instead of pme for QM/MM calculations.
After calculation, you can find the following lines:
1Link atoms algorithm: IMOMM
2Boundary:
3 QM atom 682: A hydrogen is capped at (2.484, -2.583, 3.898) Angstrom.
4 MM atom 680 has 2 bonded MM atoms: 678 681
5 MM atom 683: No MM atoms bond to it.
6 MM atom 684 has 3 bonded MM atoms: 685 686 687
7 QM atom 697: No MM atoms bond to it.
8...omitted...
9 QM atom 720: A hydrogen is capped at (-2.116, -2.240, 7.935) Angstrom.
10 MM atom 721: No MM atoms bond to it.
11 MM atom 722 has 3 bonded MM atoms: 723 724 725
12 MM atom 736 has 2 bonded MM atoms: 737 738
13 QM atom 1062: A hydrogen is capped at (-1.980, 2.584, 5.141) Angstrom.
14 MM atom 1060 has 2 bonded MM atoms: 1058 1061
15 MM atom 1063: No MM atoms bond to it.
16 MM atom 1064 has 3 bonded MM atoms: 1065 1066 1067
17...omitted...
18Total QM/MM energy
19==================
20Polarized QM energy: -1200.63363221 Hartree
21Whole system MM energy: -178.02621763 Hartree
22QM/MM energy correction: 23.67074974 Hartree
23------------------------------------------------
24QM/MM energy: -1354.98910010 Hartree
25
26Final total energy: -1354.98910010 Hartree
Lines 1-16 show how Qbics is treating the boundary atoms, the remaining lines are the QM/MM energies.
You can also visualize electronic structures with Qbics-MolStar. Load topology sys.psf in Model and coordinates sys.xyz in Coordinates, click Apply to load the solvated protein. Then, drag qmmm-2.mwfn into Qbics-MolStar to loead wavefunction. Then, right-click qmm-2.mwfn, click View Molecular Orbitals, click 91: -0.36206 (occ=2,RHF), then the HOMO of this system is shown below. The QM region is rendered using ball-and-stick representation.
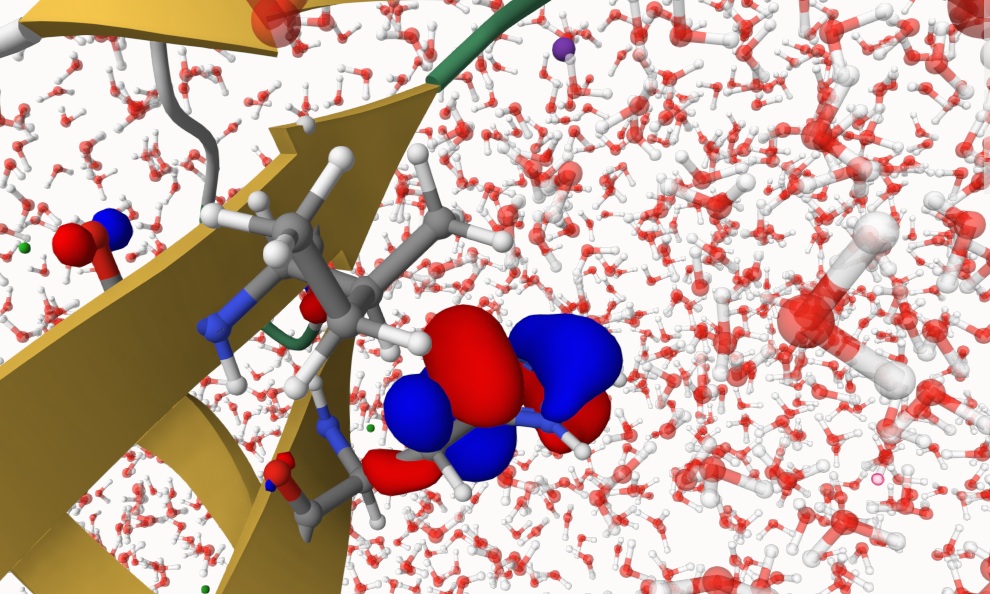
You can also visualize other properties with Qbics-MolStar.