Tip
All input files can be downloaded: Files.
scfguess
This option defines the initial guess of SCF calculations.
Options
- type
Value
hcoreWill use the eigenvectors of the core matrix as initial guess. Usually, its performance is very badatmdenWill use the superposition of converged densities of all atoms in the system as initial guess. It is recommended for most casesfragdenWill use the superposition of converged densities of fragments assigned by you as initial guess. This can be used for TSO or treating symmetry-broken systemsmwfnWill read a wave function from a MWFN file as initial guesstsoWill use a reference state (assigned withfrag) to perform TSO calculation (assigned withorb). This is only used for TSO calculationsDefault
atmdenDefine the type of initial guess. In most cases,
atmdenis recommended.mwfnwhich reads a guess from a converged SCF is also a good choice.For target state optimization (TSO), symmetry-broken, or other special calculations,
fragdencan be used.
- file
Value
A file name
Default
job name +
.mwfnWhen
typeismwfn, Qbics will read wave function with using mwfn file name.When
typeistso, Qbics will save reference wave function using this mwfn file name.
- frag
This defines atomic fragments for initial guess. The format is:
frag num_electrons spin_multiplicity atom_rangeThere can be arbitrary number of fragments, but all atoms must be included once and only once. They are only activated when
typeisfragdenortso.For example:
1scfguess 2 type fragden 3 frag 0 1 1-9 4 frag 0 -3 10-15 18 5 frag -1 3 16 17 6end
In this case, the molecule is decomposed into 3 fragments: (1) atom 1,2,3,4,5,6,7,8,9, with charge 0 and spin multiplicity 1; (2) atom 10,11,12,13,14,15,18, with charge 0 and spin multiplicity 3 (beta orbitals occupied first); (3) atom 16,17, with charge -1 and spin multiplicity 3.
For this guess, Qbics will perform 3 SCF calculations for all fragments, then superpose them as the initial guess for the SCF of the whole molecule. This can be used for ordinary, symmetry-broken, or TSO SCF.
Hint
The total charge of fragments does not have to be the same as the total system.
Hint
Please refer to the keyword
no_scfin scf for more details about TSO, when you want to usefrag.
- orb
This defines the orbitals for initial guess. The format is:
orb num_electrons spin_multiplicity alpha_MO_indices : beta_MO_indicesThere can be arbitrary number of orbital spaces, but all orbitals must be included once and only once. They are only activated when
typeismwfnortso.For example:
1scfguess 2 type mwfn 3 file x.mwfn 4 orb 12 1 1-6 : 1-6 5 orb 2 1 7 : 8 6 orb 0 1 8 : 7 7end
In this case, the orbitals will be read from
x.mwfnand 3 orbital spaces are defined: (1) alpha orbital 1,2,3,4,5,6 and beta orbital 1,2,3,4,5,6, with 12 electron and spin multiplicity 1; (2) alpha orbital 7 and beta orbital 8, with 2 electrons and spin multiplicity 1; (3) alpha orbital 8 and beta orbital 7 with 0 electron and spin multiplicity 1 (since no electrons in this orbital space, actually spin multiplicity can be arbitrary).For this guess, Qbics will read orbitals from
x.mwfnand assign occupation according toorb, then do the following ordinary or TSO SCF calculations.Also, note that in this keyword, orbital order matters. For example,
1scfguess 2 type mwfn 3 file x.mwfn 4 orb 14 1 1-6 9 7 8 10-25 : 1-6 9 7 8 10-25 5end
In this case, the 7 alpha and 7 beta electrons will occupy orbital 1,2,3,4,5,6,9.
Hint
Please refer to the keyword
no_scfin scf for more details about TSO, when you want to useorb.
- ble
This defines orbital occupations for a specific excited-state configuration. The format is:
ble block_index alpha_occ_indices : beta_occ_indicesblock_indexmust be less than the number of blocks (i.e., the number oforbcommands used to define orbital blocks).The
blekeyword is only activated whentypeis set tomwfnortso.
For example,
1scfguess 2 type mwfn 3 file x.mwfn 4 orb 12 1 1-6 : 1-6 5 orb 2 1 7-8 : 7-8 6 ble 2 1 : 2 7end
In this example:
Orbitals are read from
x.mwfn.Two orbital blocks are defined.
The
blekeyword specifies the excitation configuration:In block 2: alpha orbital 7 is occupied, and beta orbital 8 is occupied.
These correspond to occupation numbers 1 and 2, respectively.
If the
blekeyword is not used, occupations will be assigned according to the orb specification. Ifbleis used, occupations are overridden and assigned based on ble.Hint
Please refer to the keyword
no_scfin scf for more details about BLE.
Theoretical Background
XXXXXXX
Input Examples
Example: Using Initial Guess of Neutral Dieldrin for Cationic Dieldrin
In scf, we have shown hown to do SCF calculations for Dieldrin. For cationic dieldrin, the SCF took 24 cycles to converge. However, if we use the initial guess from the converged neutral dieldrin, the SCF will converge faster. To do this, we can use the following input:
1basis
2 def2-svp
3end
4
5scf
6 charge +1
7 spin2p1 2
8end
9
10scfguess
11 type mwfn
12 file scf-1a.mwfn
13end
14
15grimmedisp
16 type bj
17end
18
19mol
20 Cl 1.40900 -0.54900 -0.14200
21 C 2.91800 0.22200 -0.14800
22 C 3.34900 1.18000 -0.99000
23 Cl 2.48900 1.91800 -2.24800
24 C 4.73000 1.60600 -0.53700
25 Cl 5.24600 3.22500 -0.97500
26 C 5.80600 0.51900 -0.74700
27 C 6.83600 -0.13400 -1.70000
28 C 6.40300 -1.62200 -1.82900
29 C 6.19900 -1.81300 -0.26300
30 C 7.62200 -1.50500 0.16600
31 O 8.02000 -0.22000 0.59300
32 C 8.09500 -0.42900 -0.82100
33 C 5.11900 -0.73000 -0.15200
34 C 4.07300 -0.11600 0.82100
35 Cl 3.68500 -1.10500 2.20000
36 C 4.50000 1.37200 1.05000
37 Cl 5.90100 1.54700 2.17100
38 Cl 3.22100 2.43400 1.80000
39 H 6.52400 1.00200 -0.09500
40 H 7.05500 0.38900 -2.63100
41 H 7.19900 -2.27000 -2.21800
42 H 5.50600 -1.77200 -2.43800
43 H 5.86900 -2.82100 -0.00200
44 H 8.25600 -2.36000 0.34800
45 H 9.03900 -0.55400 -1.32700
46 H 4.42500 -1.17200 -0.89200
47end
48
49task
50 energy b3lyp
51end
Here, in scfguess...end option, we set the initial guess type to mwfn and read the wave function from scf-1a.mwfn file, which is the converged neutral dieldrin. In scfguess-1.out, you can see that the SCF will converge in 12 cycles, which is much faster than the 24 cycles without this initial guess.
Example: Superposition of Fragment Density for [CH3NH4+][HSO4-]
Now we want to calculate the energy of a cantion-anion pair [CH3NH4+][HSO4-]. The total charge of the system is 0, so the input file can be given in scfguess-2a.inp. However, chemical intuition tells us that an initial guess of superposition of CH3NH4+ and HSO4- seems to be better. This can be given in scfguess-2b.inp:
1 basis
2 def2-svp
3 end
4
5 scf
6 charge 0
7 spin2p1 1
8 end
9
10 mol
11 S -0.92132000 0.13553700 0.02022300
12 O -1.71190900 1.32700700 0.03005900
13 O -1.95455400 -1.09662200 -0.01287200
14 O -0.09369700 -0.02457400 -1.18931900
15 O -0.13508200 -0.13905500 1.21845100
16 N 2.13478700 -0.64814300 -0.03200000
17 C 3.06391300 0.49644100 0.01002200
18 H -2.74409400 -0.79280600 -0.47259900
19 H 1.37775700 -0.50028000 -0.76845000
20 H 1.50266300 -0.65123600 0.79672500
21 H 3.62254900 0.54819100 -0.92118700
22 H 3.74619000 0.39302500 0.85034900
23 H 2.46986400 1.39919200 0.12858400
24 H 2.60115300 -1.54036700 -0.14368500
25 end
26
27 task
28 energy b3lyp
29 end
1 basis
2 def2-svp
3 end
4
5 scf
6 charge 0
7 spin2p1 1
8 end
9
10 scfguess
11 type fragden
12 frag -1 1 1-5 8
13 frag +1 1 6 7 9-14
14 end
15
16 mol
17 S -0.92132000 0.13553700 0.02022300
18 O -1.71190900 1.32700700 0.03005900
19 O -1.95455400 -1.09662200 -0.01287200
20 O -0.09369700 -0.02457400 -1.18931900
21 O -0.13508200 -0.13905500 1.21845100
22 N 2.13478700 -0.64814300 -0.03200000
23 C 3.06391300 0.49644100 0.01002200
24 H -2.74409400 -0.79280600 -0.47259900
25 H 1.37775700 -0.50028000 -0.76845000
26 H 1.50266300 -0.65123600 0.79672500
27 H 3.62254900 0.54819100 -0.92118700
28 H 3.74619000 0.39302500 0.85034900
29 H 2.46986400 1.39919200 0.12858400
30 H 2.60115300 -1.54036700 -0.14368500
31 end
32
33 task
34 energy b3lyp
35 end
In scfguess-2b.inp, the fragment initial guess is set using
1 scfguess
2 type fragden
3 frag -1 1 1-5 8
4 frag +1 1 6 7 9-14
5 end
Here, the atomic indices of CH3NH4+ and HSO4- are given in 1-5 8 and 6 7 9-14, respectively, and charges and spin multiplicities are also given. The atomic indices are shown below:
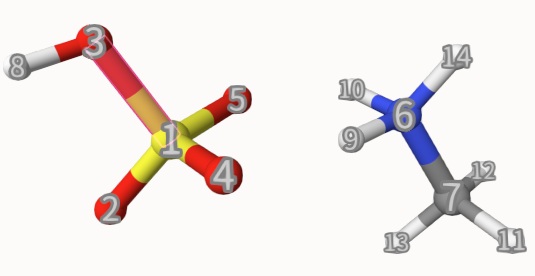
In their output scfguess-2a.out and scfguess-2b.out, both give the same energy, but the SCF cycles are 25 and 11, respectively. Thus, superposition of fragments is indeed a better initial guess.
Also, we can check the Mulliken population of this system:
1Mulliken Populations
2====================
3 # Symbol Charge Spin
4----------------------------------------------
5 1 S 1.06432061 0.00000000
6 2 O -0.48154427 0.00000000
7 3 O -0.35593323 0.00000000
8 4 O -0.56975508 0.00000000
9 5 O -0.56231274 0.00000000
10 6 N -0.03632083 0.00000000
11 7 C 0.05819466 0.00000000
12 8 H 0.18495781 0.00000000
13 9 H 0.18311761 0.00000000
14 10 H 0.17532487 0.00000000
15 11 H 0.05607280 0.00000000
16 12 H 0.05428454 0.00000000
17 13 H 0.07504981 0.00000000
18 14 H 0.15454343 0.00000000
19----------------------------------------------
20 Sum -0.00000000 0.00000000
21----------------------------------------------
We can see that, the sum of Mulliken charge on CH3NH4+ and HSO4- are +0.72 and -0.72, respectively, being quite different from their ideal value +1 and -1. This is actually the electron delocalization effect.
Example: Diatatic State of [CH3NH4+][HSO4-] with TSO-DFT
Tip
For a complete tutorial of TSO-DFT, please refer to:
In the last example, we have calculated [CH3NH4+][HSO4-], where electron is delocalzied over the entire cluster. Can we calculate a state where all electrons are strictly localzied on each molecule? This can be done easily with Qbics. Such state can be called a diabatic state. Diabatic state can be calculated with TSO-DFT method, which can be done with scfguess-3.inp:
1 basis
2 def2-svp
3 end
4
5 scf
6 charge 0
7 spin2p1 1
8 no_scf tso
9 type U # This must be given for TSO calculation.
10 end
11
12 scfguess
13 type fragden
14 frag -1 1 1-5 8
15 frag +1 1 6 7 9-14
16 end
17
18 mol
19 S -0.92132000 0.13553700 0.02022300
20 O -1.71190900 1.32700700 0.03005900
21 O -1.95455400 -1.09662200 -0.01287200
22 O -0.09369700 -0.02457400 -1.18931900
23 O -0.13508200 -0.13905500 1.21845100
24 N 2.13478700 -0.64814300 -0.03200000
25 C 3.06391300 0.49644100 0.01002200
26 H -2.74409400 -0.79280600 -0.47259900
27 H 1.37775700 -0.50028000 -0.76845000
28 H 1.50266300 -0.65123600 0.79672500
29 H 3.62254900 0.54819100 -0.92118700
30 H 3.74619000 0.39302500 0.85034900
31 H 2.46986400 1.39919200 0.12858400
32 H 2.60115300 -1.54036700 -0.14368500
33 end
34
35 task
36 energy b3lyp
37 end
We just need to add no_scf tso to scf...end, and give fragment definition in scfguess...end. Also, you must explicitly write type U to enforce unrestricted SCF. Such method can be called TSO-B3LYP. In the output scfguess-3.out:
1Mulliken Populations
2====================
3 # Symbol Charge Spin
4----------------------------------------------
5 1 S 1.05819195 -0.00000000
6 2 O -0.49810728 0.00000000
7 3 O -0.36249350 -0.00000000
8 4 O -0.71399133 0.00000000
9 5 O -0.66324621 -0.00000000
10 6 N -0.09172438 -0.00000000
11 7 C 0.05015632 0.00000000
12 8 H 0.17964637 0.00000000
13 9 H 0.35411645 0.00000000
14 10 H 0.29719708 0.00000000
15 11 H 0.06307774 -0.00000000
16 12 H 0.06123412 -0.00000000
17 13 H 0.09862277 -0.00000000
18 14 H 0.16731989 0.00000000
19----------------------------------------------
20 Sum -0.00000000 -0.00000000
21----------------------------------------------
22..omitted..
23Final total energy: -795.63743617 Hartree
We can see that, the sum of Mulliken charge on CH3NH4+ and HSO4- are +1 and -1. So we have successfully obtained the diabatic state we need. Also, its energy -795.63743617 Hartree is higher than the one obatained from scfguess-2b.out: -795.67538949 Hartree. Their difference
-795.67538949 Hartree--795.63743617 Hartree = 23.82 kcal/mol
is actually the Charge transfer interaction energy without BSSE correction defined in TSO-EDA, which can be calculated with eda. The following input:
1basis
2 def2-svp
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type U # This must be given.
9end
10
11eda
12 type tso
13 frag -1 1 1-5 8
14 frag +1 1 6 7 9-14
15end
16
17mol
18 S -0.92132000 0.13553700 0.02022300
19 O -1.71190900 1.32700700 0.03005900
20 O -1.95455400 -1.09662200 -0.01287200
21 O -0.09369700 -0.02457400 -1.18931900
22 O -0.13508200 -0.13905500 1.21845100
23 N 2.13478700 -0.64814300 -0.03200000
24 C 3.06391300 0.49644100 0.01002200
25 H -2.74409400 -0.79280600 -0.47259900
26 H 1.37775700 -0.50028000 -0.76845000
27 H 1.50266300 -0.65123600 0.79672500
28 H 3.62254900 0.54819100 -0.92118700
29 H 3.74619000 0.39302500 0.85034900
30 H 2.46986400 1.39919200 0.12858400
31 H 2.60115300 -1.54036700 -0.14368500
32end
33
34task
35 eda b3lyp
36end
At the end of the output scfguess-3eda.out:
1Target State Optimized Wavefunction Energy Decomposition Analysis
2=================================================================
3WITHOUT BSSE correction:
4Electrostatic interaction energy: -129.90 kcal/mol
5Exchange-correlation interaction energy: 35.20 kcal/mol
6Polarization interaction energy: -13.18 kcal/mol
7Charge transfer interaction energy: -23.82 kcal/mol
8Grimme's dispersion interaction: 0.00 kcal/mol
9----------------------------------------------------------------
10Total interaction energy: -131.69 kcal/mol
11
12WITH BSSE correction:
13Electrostatic interaction energy: -129.90 kcal/mol
14Exchange-correlation interaction energy: 35.20 kcal/mol
15Polarization interaction energy: -13.18 kcal/mol
16Charge transfer interaction energy: -16.31 kcal/mol
17Grimme's dispersion interaction: 0.00 kcal/mol
18----------------------------------------------------------------
19Total interaction energy: -124.18 kcal/mol
Line 7 says Charge transfer interaction energy: -23.82 kcal/mol, which is exactly the same value as we calculated.
Example: TSO-DFT for Doubly Excited States of HCHO
Tip
For a complete tutorial of TSO-DFT, please refer to:
As mentioned above, TSO-DFT can be used to study diabatic states. It can also be used to study excited states. Here, we demonstrate how to use TSO-DFT to calculate a doubly excitated state of HCHO. We consider HOMO2 → LUMO2:
1basis
2 element
3 H cc-pVTZ
4 C cc-pCVTZ
5 O cc-pCVTZ
6end
7
8scf
9 charge 0
10 spin2p1 1
11 type U # For TSO-DFT, unrestricted SCF is preferred.
12 no_scf TSO
13end
14
15scfguess
16 type tso
17 file hcho-ref.mwfn
18 frag 0 1 1-4
19 orb 16 1 1-7 9-114 : 1-7 9-114
20 orb 0 1 8 : 8
21end
22
23mol
24 C -0.000756 -0.520733 0.
25 H 0.935697 -1.111766 0.
26 H -0.939631 -1.107897 0.
27 O 0.001792 0.678123 0
28end
29
30task
31 energy b3lyp
32end
Here:
Line 16: Set
typetotsoenable special initial guess for TSO-DFT;Line 17: The reference wavefunction (ground state of HCHO) is saved to
hcho-ref.mwfn;Line 18: Define the reference state. Here, the whole molecule is treated as the reference state;
Line 19,20: Define the orbital spaces to set up the required excited state as explained above.
After calculation, you can find these lines in scfguess-4a.out:
1TSO Transition
2==============
3Reference wave function read from: hcho-ref.mwfn
4Reference energy: -114.55175800 Hartree
5Current energy: -114.15907196 Hartree
6E(Current)-E(Ref): 10.68553145 eV
7Transition dipole moment (Debye): 0.00000 0.00000 0.00000
8Oscillator strength: 0.00000
9Higher order corrections:
10Transition quadrupole moment (Debye*Angstrom):
11 Qxx: 0.00000; Qyy: 0.00000; Qzz: 0.00000
12 Qxy: 0.00000; Qxz: 0.00000; Qyz: 0.00000
13Quadrupole correction to oscillator strength: 0.00000E+00
14Transition angular momentum (au): 0.00000 0.00000 0.00000
15Magnetic dipole correction to oscillator strength: 0.00000E+00
16
17 ---- Self Consistent Field Energy Done ------------------
18
19Final total energy: -114.15907196 Hartree
The double excitation energy is 10.68 eV, which agrees well with the one obtained from EOM-CC (10.34 eV). Note that, by changing energy in scfguess-4a.inp to opt, you can do geometry optimization for this excited state!
You can also use an MWFN file as the initial guess to do TSO-DFT calculation:
1basis
2 element
3 H cc-pVTZ
4 C cc-pCVTZ
5 O cc-pCVTZ
6end
7
8scf
9 charge 0
10 spin2p1 1
11 type U # For TSO-DFT, unrestricted SCF is preferred.
12 no_scf TSO
13end
14
15scfguess
16 type mwfn
17 file hcho-ref.mwfn
18 orb 16 1 1-7 9-114 : 1-7 9-114
19 orb 0 1 8 : 8
20end
21
22mol
23 C -0.000756 -0.520733 0.
24 H 0.935697 -1.111766 0.
25 H -0.939631 -1.107897 0.
26 O 0.001792 0.678123 0
27end
28
29task
30 energy b3lyp
31end
Note that we have deleted frag 0 1 1-4 since reference wavefucntion is already given in hcho-ref.mwfn. The obtained energy in scfguess-4b.out is exactly the same as in scfguess-4a.out.