Tip
All input files can be downloaded: Files.
Tip
For more information of this section, please refer to these pages:
Density Functional Theory Calculations
This tutorial will lead you step by step to do density functional theory (DFT) calculations using Qbics. Here we only do energy calculations. For geometry optimization and molecular dynamics, please refer to Geometry Optimization and Standard Molecular Dynamics Simulations.
Example: Single Point Energy of Water
We will now do the first calculation using Qbics. The following input file will calculate B3LYP-D3BJ/def2-tzvp energy for a water molecule:
1# This first calculation.
2
3basis
4 def2-tzvp
5end
6
7scf
8 charge 0
9 spin2p1 1
10 type R # You do not have to write it. The program will determine it by itself.
11end
12
13grimmedisp
14 type bj
15end
16
17mol
18 O 0. 0.00000000 -0.11081188
19 H 0. -0.78397589 0.44324751
20 H 0. 0.78397589 0.44324751
21end
22
23task
24 energy b3lyp
25end
Then run it:
$ qbics water-1.inp -n 4 > water-1.out
Here, -n 4 means Qbics will use 4 CPU cores for parallization. This calculation is very fast. After calculation, you will find 2 new files:
water-1.out water-1.mwfn
water-1.out is the output file for this calculation. You can find energies, molecular orbital (MO) information, spin, population, electric multipole moments in it:
1SCF Energies
2============
3Kinetic energy: 76.16685643 Hartree
4Electron-nuclear attraction energy: -199.22128800 Hartree
5Pseudopotential energy: 0.00000000 Hartree
6Exchange-correlation energy: -7.55662337 Hartree
7Electron Coulomb energy: 46.77766964 Hartree
8Electron exchange energy: -1.78638759 Hartree
9Nuclear repulsion energy: 9.15711600 Hartree
10Grimme dispersion energy: -0.00057358 Hartree
11----------------------------------------------------------------
12SCF energy (E): -76.46323045 Hartree
13Virial quotien (V/T): -2.00389112
14
15Molecular Orbitals
16==================
17k = Gamma
18HOMO-LUMO (5-6) gap: 8.990 eV
19 # Occupancies Energies/Hartree
20 1 2.000 -19.12516259
21 2 2.000 -1.01223986
22 3 2.000 -0.54372159
23 4 2.000 -0.38293836
24...
25Spin
26====
27Expected <S^2>: 0.00000; S = 0.00000
28Calculated <S^2>: 0.00000; S = 0.00000
29
30Mulliken Populations
31====================
32 # Symbol Charge Spin
33----------------------------------------------
34 1 O -0.64331377 0.00000000
35 2 H 0.32165689 0.00000000
36 3 H 0.32165689 0.00000000
37----------------------------------------------
38 Sum -0.00000000 0.00000000
39----------------------------------------------
40
41Electric Multipole Moments
42==========================
43 # Total Electronic Nuclear Unit
44------------------------------------------------------------------------------------
45Charge:
46 0 -0.0000 -10.0000 10.0000 |e|
47Dipole moment:
48 X -0.0000 -0.0000 0.0000 Debye
49 Y 0.0000 0.0000 0.0000 Debye
50 Z 1.9936 1.9936 -0.0000 Debye
51 Totla 1.9936 Debye
52Quadrupole moment:
53 XX -7.5861 -7.5861 0.0000 Debye*Angstrom
54 XY -0.0000 -0.0000 0.0000 Debye*Angstrom
55 XZ -0.0000 -0.0000 0.0000 Debye*Angstrom
56 YY -4.1639 -10.0682 5.9043 Debye*Angstrom
57 YZ 0.0000 0.0000 -0.0000 Debye*Angstrom
58 ZZ -6.4570 -8.8162 2.3592 Debye*Angstrom
59------------------------------------------------------------------------------------
water-1.mwfn is the wave function file in Multiwfn format. You can do all wave function visualization and analysis with Multiwfn. A faster way is to use Qbics-MolStar, an online app, you can also download it on your own Windows machine.
Just drag water-1.mwfn into explorer, and it will be automatically loaded. Right-click water-1.mwfn and select View Molecular Orbitals:
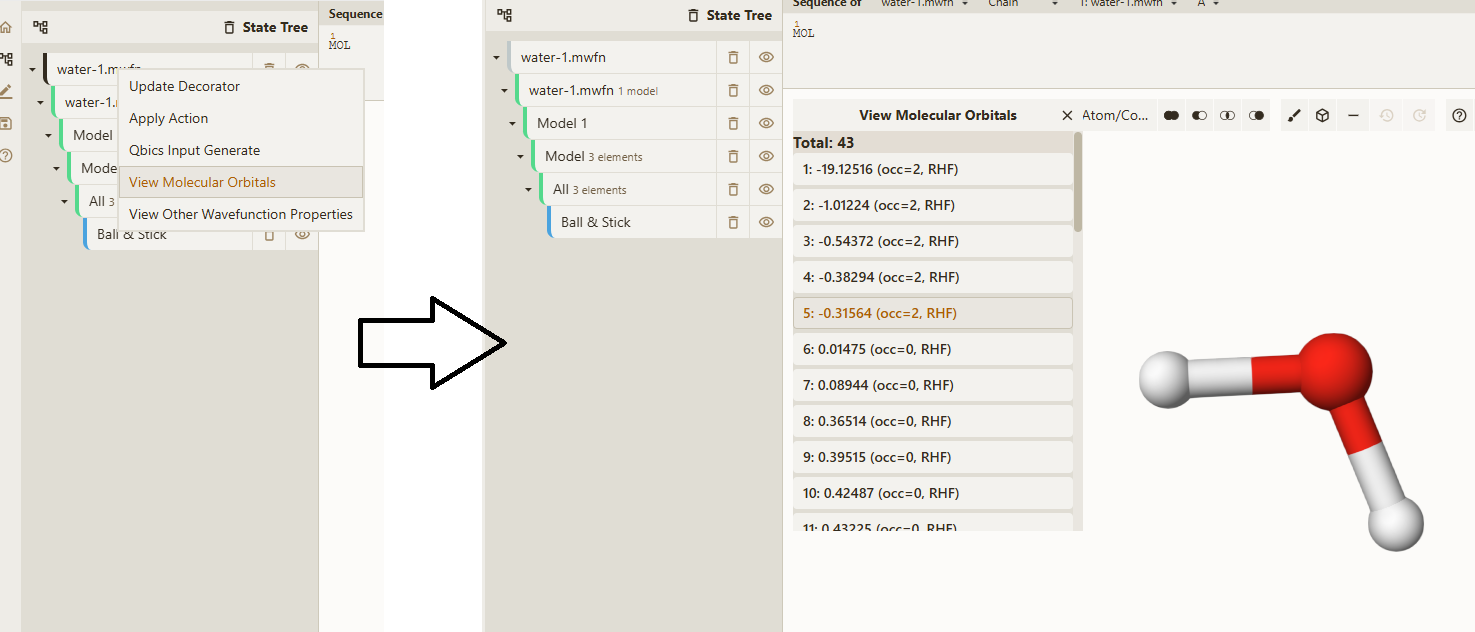
Now, select 5: -0.313564 (occ=2, RHF), which is the HOMO of water, then MO will be rendered:
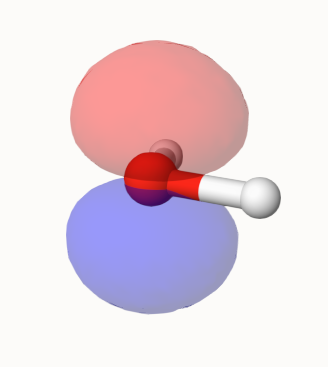
You can also view other MOs or properties.
Now we will explain the input water-1.inp of the last example.
Anything after
#is treated as comments. You can write anything anywhere in the input file.basis ... endThis block defines the basis set used the calculation. You can find all available basis sets inqbics/basis. Just input the file name. You can also define your own basis set, or set basis set for different elements. See below.scf ... endThis block controls how the self-consistent field (SCF) calculations are done. By its name, you may understand that:
chargeThe charge of the molecule, like0,+3,-1, etc.
spin2p1The spin multiplicity. For a molecule with \(n\) unpaired electrons, this total spin is \(S=\frac{n}{2}\), so spin multiplicity is \(2S+1 = n+1\). For example, for a triplet state of water, you will usespin2p1 3, then 2 unpaired electrons will occupy 2 alpha orbitals.
typeThe value isRif a restricted Hartree–Fock (HF) or Kohn–Sham (KS) is needed, orUis an unrestricted one is needed. If you do not write this, the program will determine it automatically according to spin multiplicity:Rfor singlet andUfor other cases
grimmedisp ... endThis block controls how Grimme dispersion correction is applied. Here we usebjto indicate Becke–Johnson dampling D3 correction.mol ... endThis block gives the molecule coordinates in XYZ format. You can also simply give a coordinate file name in XYZ or PDB format, saywater.xyzorwater.pdb.task ... endThis tells Qbics what it should do.energy b3lypmeans it will use B3LYP to calculate energy. You can put several tasks in this block.
Based on the explanations above, you can now try to calculate the triplet state of water:
1# Triplet state of water.
2
3basis
4 def2-tzvp
5end
6
7scf
8 charge 0
9 spin2p1 3
10 type U # If you do not write it. The program will determine it by itself.
11end
12
13grimmedisp
14 type bj
15end
16
17mol
18 O 0. 0.00000000 -0.11081188
19 H 0. -0.78397589 0.44324751
20 H 0. 0.78397589 0.44324751
21end
22
23task
24 energy b3lyp
25end
You can find MO occupations and spin here:
1Molecular Orbitals
2==================
3k = Gamma
4Alpha HOMO-LUMO (6-7) gap: 3.247 eV
5Beta HOMO-LUMO (4-5) gap: 5.963 eV
6 Alpha Alpha Beta Beta
7 # Occupancies Energies/Hartree Occupancies Energies/Hartree
8 1 1.000 -19.35044069 1.000 -19.31689276
9 2 1.000 -1.21472533 1.000 -1.12845421
10 3 1.000 -0.69876887 1.000 -0.66807740
11 4 1.000 -0.57036857 1.000 -0.52597568
12 5 1.000 -0.56380445 0.000 -0.30683178
13 6 1.000 -0.10311895 0.000 -0.01035402
14...
15Spin
16====
17Expected <S^2>: 2.00000; S = 1.00000
18Calculated <S^2>: 2.00188; S = 1.00063
The spin of this calculation is 1.00063, which is very close to the theoretical value 1, indicating a very small spin contamination.
More Basis Set Configurations
Qbics hase provided flexible basis set configuration. This section will show it.
Define Your Own Basis Set
Assume you want to use a basis set called pc-2 for water. However, this is not available in qbics/basis, you can then define it by your self.
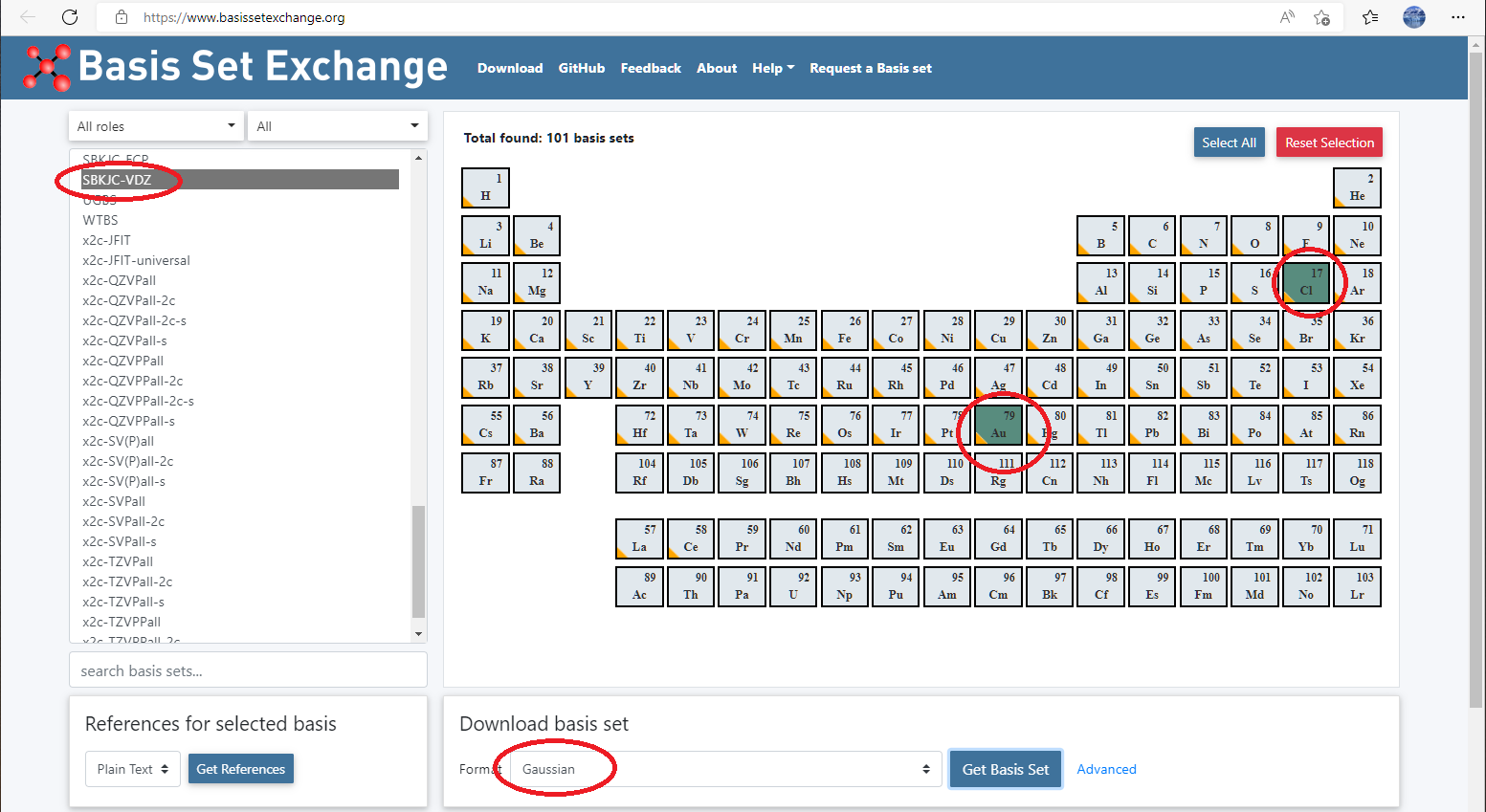
Go to basis set exchange website: https://www.basissetexchange.org/, choose
pc-2for H and O, and download it using GAUSSIAN format.
Attention
Replace all D to E in the basis set definitions!
Now we have two ways:
The first way: Simply copy it to
basis ... endblock:
1# This first calculation.
2
3basis
4 # From: https://www.basissetexchange.org/basis/pc-2/format/gaussian94/?version=0& elements=1,8
5 H 0
6 S 4 1.00
7 0.754230E+02 0.240650E-02
8 0.113500E+02 0.184870E-01
9 0.259930E+01 0.897420E-01
10 0.735130E+00 0.281110E+00
11 S 1 1.00
12 0.231670E+00 0.100000E+01
13 S 1 1.00
14 0.741470E-01 0.100000E+01
15 P 1 1.00
16 0.160000E+01 0.100000E+01
17 P 1 1.00
18 0.450000E+00 0.100000E+01
19 D 1 1.00
20 0.125000E+01 0.100000E+01
21 ****
22 O 0
23 S 7 1.00
24 0.147820E+05 0.535190E-03
25 0.221730E+04 0.413750E-02
26 0.504740E+03 0.212450E-01
27 0.142870E+03 0.824530E-01
28 0.463000E+02 0.236710E+00
29 0.163370E+02 0.440390E+00
30 0.598280E+01 0.364650E+00
31 S 7 1.00
32 0.221730E+04 -0.192750E-05
33 0.504740E+03 -0.579640E-04
34 0.142870E+03 -0.794940E-03
35 0.463000E+02 -0.731250E-02
36 0.163370E+02 -0.405740E-01
37 0.598280E+01 -0.915940E-01
38 0.167180E+01 0.209400E+00
39 S 1 1.00
40 0.646620E+00 0.100000E+01
41 S 1 1.00
42 0.216690E+00 0.100000E+01
43 P 4 1.00
44 0.604240E+02 0.689490E-02
45 0.139350E+02 0.490050E-01
46 0.415310E+01 0.182550E+00
47 0.141580E+01 0.376330E+00
48 P 1 1.00
49 0.475490E+00 0.100000E+01
50 P 1 1.00
51 0.145290E+00 0.100000E+01
52 D 1 1.00
53 0.220000E+01 0.100000E+01
54 D 1 1.00
55 0.650000E+00 0.100000E+01
56 F 1 1.00
57 0.110000E+01 0.100000E+01
58 ****
59end
60
61scf
62 charge 0
63 spin2p1 1
64 type R # You need not to write it. The program will determine it by itself.
65end
66
67grimmedisp
68 type bj
69end
70
71mol
72 O 0. 0.00000000 -0.11081188
73 H 0. -0.78397589 0.44324751
74 H 0. 0.78397589 0.44324751
75end
76
77task
78 energy b3lyp
79end
Then do the calculation as usual.
The second way: Save the basis set in a file called, say,
pc-2-OH.txt. Assume you put it in/home/you/calc/pc-2-OH.txt, then write this file name inbasis ... endblock:
1# This first calculation.
2
3basis
4 /home/you/calc/pc-2-OH.txt
5end
6
7scf
8 charge 0
9 spin2p1 1
10 type R # You need not to write it. The program will determine it by itself.
11end
12
13grimmedisp
14 type bj
15end
16
17mol
18 O 0. 0.00000000 -0.11081188
19 H 0. -0.78397589 0.44324751
20 H 0. 0.78397589 0.44324751
21end
22
23task
24 energy b3lyp
25end
Both ways will give the same result.
Different Basis Sets for Different Elements
Assume you want to use pc-2 (defined above) for oxygen but cc-pvdz for hydrogen, you can use the following basis ... end:
1basis
2 element # This indicates that Qbics will assign basis set element by element.
3 O /home/you/calc/pc-2-OH.txt
4 H cc-pvdz
5end
The word element in the first line of basis ... end block means that Qbics will assign basis set element by element. Then simply write the basis set for every element.
Example: Heavy Elements with Pseudopotential
General Rules
For heavy elements, one should use pseudopotential for reasonable calculations.
Attention
You must write BOTH valence basis set and pseudopotential in the input file. If pseudopotential is not written explicitly in the input file, Qbics will NOT use it.
This is because for some elements, there exist both all-electron basis set and valence-only basis set with pseudopotential at the same time, so you should make it clear in the input file.
The valence basis sets and core pseudopotentials are stored in qbics/basis and qbics/pseudopotential, respectively. For an element, you must use them consistently:
Valence Basis Set |
Pseudopotential |
|---|---|
def2-X |
def2-ecp |
(aug-)cc-X-pp |
cc-ecp |
lanlX |
lanl-ecp |
For example, you can use def2-TZVPP and def-ecp for any element, but NEVER use def2-TZVPP and lanl-ecp together!
Assume you want to calculate B3LYP-D3BJ energy for AuCl3, you can use def2- series:
1basis
2 def2-tzvp
3end
4
5pseudopotential
6 def2-ecp
7end
8
9scf
10 charge 0
11 spin2p1 1
12end
13
14grimmedisp
15 type bj
16end
17
18mol
19 Au 0.00000000 0.00000000 0.
20 Cl 0.00000000 -2.33000000 0.
21 Cl 2.01783919 1.16500000 0.
22 Cl -2.01783919 1.16500000 0.
23end
24
25task
26 energy b3lyp
27end
Or, you may want to use cc-pvdz for chlorine and cc-pvdz-pp for gold, then the input is:
1basis
2 element
3 Cl cc-pvdz
4 Au cc-pvdz-pp
5end
6
7pseudopotential
8 cc-ecp
9end
This means that for the valence and core part of gold, cc-pvdz basis set and pseudopotential is used, respectively.
Define Your Own Pseudopotential
You can also define your own pseudopotential like we have done for basis set. Again for AuCl3, we want to use SBKJC valence basis set and pseudopotentials, then
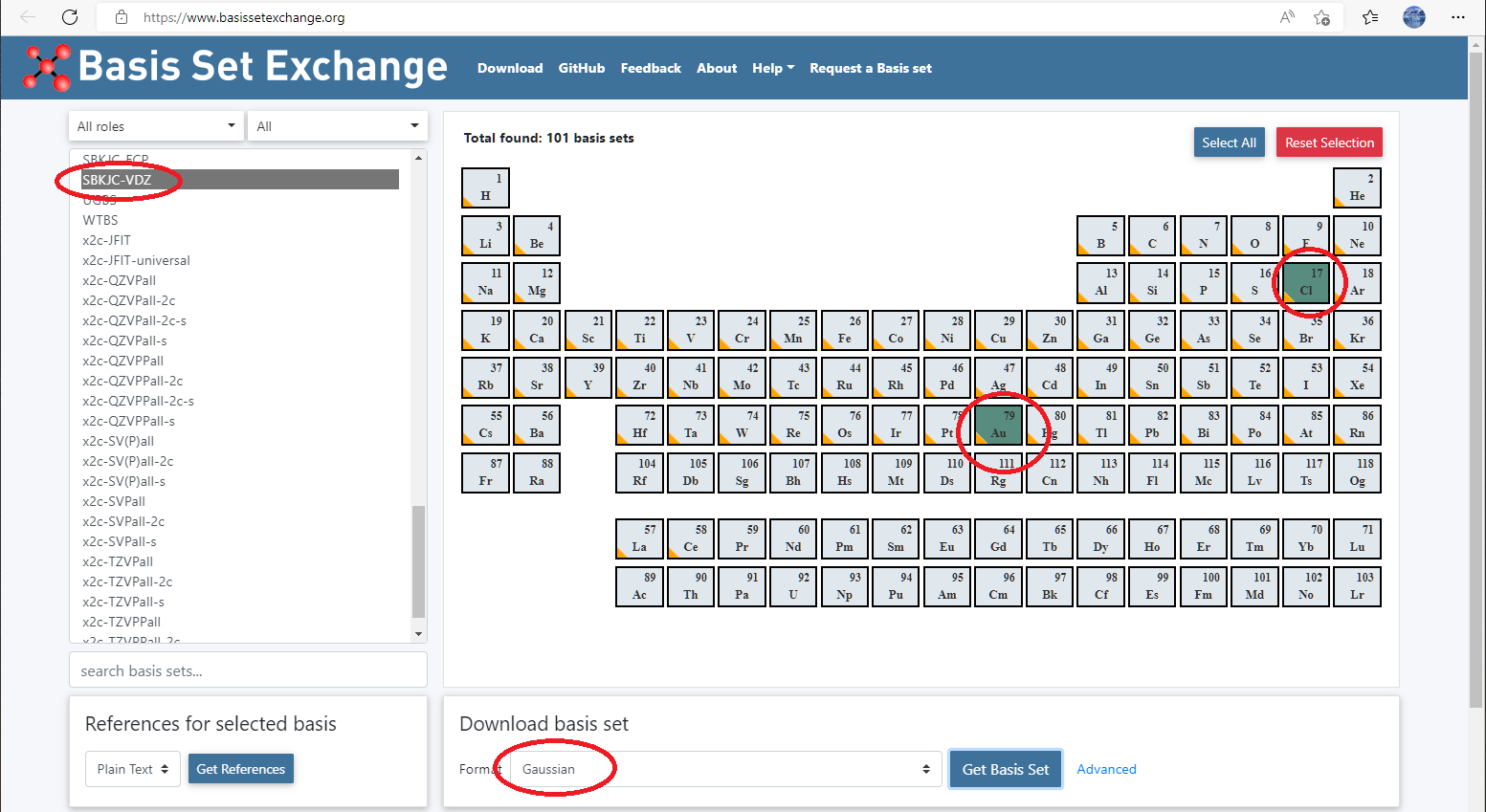
Go to basis set exchange website: https://www.basissetexchange.org/, choose
SBKJC-VDZfor Au and Cl, and download it using GAUSSIAN format.
Paste basis set and pseudotential definitions in
basis ... endandpseudopotential ... endblock, respectively:
1basis
2 # From: https://www.basissetexchange.org/basis/sbkjc-vdz/format/gaussian94/?version=0&elements=17,79
3 Cl 0
4 SP 3 1.00
5 2.225000 -.330980 -.126040
6 1.173000 0.115280 0.299520
7 0.385100 0.847170 0.583570
8 SP 1 1.00
9 0.130100 0.265340 0.340970
10 ****
11 Au 0
12 SP 1 1.00
13 1.502000 1.000000 1.000000
14 SP 4 1.00
15 7.419000 0.222546 0.019924
16 4.023000 -1.086045 -.299997
17 1.698000 1.156039 0.748919
18 0.627100 0.518061 0.504023
19 SP 1 1.00
20 0.151500 1.000000 1.000000
21 SP 1 1.00
22 0.049250 1.000000 1.000000
23 D 3 1.00
24 3.630000 -.087402
25 1.912000 0.468634
26 0.842300 0.654805
27 D 1 1.00
28 0.375600 1.000000
29 D 1 1.00
30 0.154400 1.000000
31 ****
32end
33
34pseudopotential
35 # From: https://www.basissetexchange.org/basis/sbkjc-vdz/format/gaussian94/?version=0&elements=17,79
36 CL 0
37 CL-ECP 2 10
38 d potential
39 1
40 1 4.8748300 -3.4073800
41 s-d potential
42 2
43 0 17.0036700 6.5096600
44 2 4.1038000 42.2778500
45 p-d potential
46 2
47 0 8.9002900 3.4286000
48 2 3.5264800 22.1525600
49 ****
50 AU 0
51 AU-ECP 4 60
52 g potential
53 1
54 1 4.3876300 -10.7235800
55 s-g potential
56 3
57 0 1.5563600 6.3561200
58 2 3.7159300 -364.4403900
59 2 4.0679200 428.1975300
60 p-g potential
61 3
62 0 1.1879800 4.4151800
63 2 3.0155100 -136.5755000
64 2 3.5958800 194.2053500
65 d-g potential
66 2
67 0 35.2500000 8.8819800
68 2 5.0230700 86.7661200
69 f-g potential
70 1
71 0 1.6888100 6.2160300
72 ****
73end
74
75scf
76 charge 0
77 spin2p1 1
78end
79
80grimmedisp
81 type bj
82end
83
84mol
85 Au 0.00000000 0.00000000 0.
86 Cl 0.00000000 -2.33000000 0.
87 Cl 2.01783919 1.16500000 0.
88 Cl -2.01783919 1.16500000 0.
89end
90
91task
92 energy b3lyp
93end
Note, you MUST add **** between pseudopential definitions for each element.
Or, you can save both definitions in
/home/you/calc/SBJKC.txtand/home/you/calc/SBJKC-ECP.txt, respectively, and write these file names in the input file:
1basis
2 /home/you/calc/SBJKC.txt
3end
4
5pseudopotential
6 /home/you/calc/SBJKC-ECP.txt
7end
Example: Set Special SCF Initial Guess
Qbics supports several ways to set SCF initial guess. The default one is the “superposition of atomic densities”, which works quiet well in most cases. Sometimes, one may or must use other ways.
Guess From Another Calculations
Consider CH3NH3+OH-, we do a B3LYP-D3BJ/6-31g(d) calculation:
1basis
2 6-31g(d)
3end
4
5scf
6 charge 0
7 spin2p1 1
8end
9
10grimmedisp
11 type bj
12end
13
14mol
15C -0.02909313 -0.38166253 -1.63963825
16H 0.82173570 -1.03039025 -1.62769450
17H -0.03691367 0.17958863 -2.55059158
18H -0.92347728 -0.96516294 -1.57252437
19N 0.04484969 0.58404262 -0.44233044
20H 0.93923384 1.16754303 -0.50944432
21H -0.80597913 1.23277035 -0.45427419
22H 0.05267023 0.02279146 0.46862289
23O 0.06914388 -0.26694332 2.08310089
24H -0.31212699 -0.12725829 2.95299779
25end
26
27task
28 energy b3lyp
29end
After calculation, we will obtain a file ch3nh3oh-1.mwfn. We can use this as an initial guess for a more expensive calculation, like B3LYP-D3BJ/6-311g(2df,2pd) calculation:
1basis
2 6-311g(2df,2pd)
3end
4
5scf
6 charge 0
7 spin2p1 1
8end
9
10scfguess
11 type mwfn
12 file ch3nh3oh-1.mwfn
13end
14
15grimmedisp
16 type bj
17end
18
19mol
20C -0.02909313 -0.38166253 -1.63963825
21H 0.82173570 -1.03039025 -1.62769450
22H -0.03691367 0.17958863 -2.55059158
23H -0.92347728 -0.96516294 -1.57252437
24N 0.04484969 0.58404262 -0.44233044
25H 0.93923384 1.16754303 -0.50944432
26H -0.80597913 1.23277035 -0.45427419
27H 0.05267023 0.02279146 0.46862289
28O 0.06914388 -0.26694332 2.08310089
29H -0.31212699 -0.12725829 2.95299779
30end
31
32task
33 energy b3lyp
34end
Here, scfguess ... end block controls the initial guess with type, which can be:
Option |
Meaning |
|---|---|
|
Diagonalization of core Hamiltonian. NOT recommended. |
|
Superposition of atomic densities. Default. |
|
Read a wave function from a mwfn file given by |
|
Superposition of molecular fragment densities. See below. |
Attention
For an input file x.inp, the program will rewrite x.mwfn during the calculation, so if you want to keep the x.mwfn, you can copy it to something like x-1.mwfn, and write
scfguess
type mwfn
file x-1.mwfn
end
Then the program will read x-1.mwfn for initial guess but not overwrite x.mwfn.
Guess From Fragment Superposition
Consider CH3NH3+OH-, it seems to be logical that one can use the superposition of CH3NH3+ and OH- as initial guess. This is very easy:
1basis
2 6-311g(2df,2pd)
3end
4
5scf
6 charge 0
7 spin2p1 1
8end
9
10scfguess
11 type fragden
12 frag +1 1 1-8
13 frag -1 1 9 10
14end
15
16grimmedisp
17 type bj
18end
19
20mol
21C -0.02909313 -0.38166253 -1.63963825
22H 0.82173570 -1.03039025 -1.62769450
23H -0.03691367 0.17958863 -2.55059158
24H -0.92347728 -0.96516294 -1.57252437
25N 0.04484969 0.58404262 -0.44233044
26H 0.93923384 1.16754303 -0.50944432
27H -0.80597913 1.23277035 -0.45427419
28H 0.05267023 0.02279146 0.46862289
29O 0.06914388 -0.26694332 2.08310089
30H -0.31212699 -0.12725829 2.95299779
31end
32
33task
34 energy b3lyp
35end
Here, type fragden means we will use superposition of fragments as initial guess. The fragments can be defined with frag:
frag +1 1 1-8
The first and second number is the charge and spin multiplicity of the fragment. Then is the atom indices for this fargment. It can be discontinuous like:
frag +1 1 1 2 5 8-12 15
In this case, the atom 1 2 5 8 9 10 11 12 15 will be a fragment. You can define any numbers of fragments.
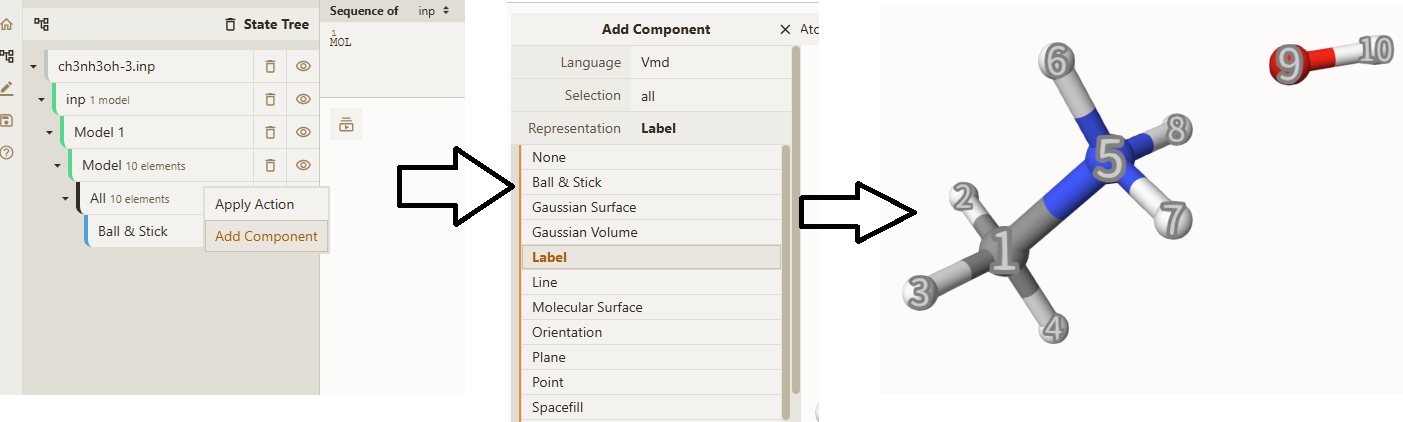
To determine the atomic indices, it is very convienent to use Qbics-MolStar. Just drag ch3nh3oh-3.inp into explore, right-click All, then click Add Component, then click Label in Representation, then click + Create Component, you can see clearly the atomic indices:
SCF Convergence Condition
There are 3 options in scf ... end block to control the convergence condition:
scf
energy_cov 1.E-6
density_cov 1.E-6
max_it 100
end
Here, energy_cov and density_cov means that the SCF is thought to converge when the energy and density change is smaller than 1.E-6 and 1.E-6, respectively. max_it means the maximum number of SCF iterations. You can change these when you need.